Нарушения сократимости и насосной функции сердца

Механизмы компенсации гемодинамических нарушений при острой сердечной недостаточности. На начальной стадии систолической дисфункции желудочков сердца включаются интракардиальные факторы компенсации сердечной недостаточности, важнейшим из которых является механизм Франка-Старлинга (гетерометрический механизм компенсации, гетерометрическая гиперфункция сердца). Реализацию его можно представить… Читать ещё >
Нарушения сократимости и насосной функции сердца (реферат, курсовая, диплом, контрольная)
Сердечная недостаточность — неспособность сердца выполнять насосную функцию вследствие существенного снижения сократительной способности миокарда, а также поражения клапанов сердца или пороков развития системы кровообращения.
К основным причинам развития сердечной недостаточности относятся: 1) первичное поражение миокарда, приводящее к нарушению его сократимости. Возникает при ИБС (постинфарктный и атеросклеротический кардиосклероз), дилатационной кардиомиопатии, миокардитах, миокардиодистрофиях; 2) перегрузка давлением в фазу систолы. Это нарушение характеризуется увеличением работы сердца (например, при артериальной гипертензии или аортальном стенозе; 3) перегрузка объемом в фазу диастолы, сопровождающаяся увеличением работы сердца при аортальной или митральной недостаточности, дефекте межжелудочковой перегородки; 4) снижение наполнения желудочков (преимущественно диастолическая недостаточность). Развивается при гипертрофической кардиомиопатии, гипертоническом сердце (при отсутствии дилатации левого желудочка), изолированном митральном стенозе, констриктивном и экссудативном перикардите; 5) высокий сердечный выброс (при тиреотоксикозе, выраженной анемии и т. д.).
Классификация сердечной недостаточности. В зависимости от происхождения выделяют следующие формы сердечной недостаточности:
- 1) миокардиальная, которая обусловлена первичным поражением мышцы сердца физическими, химическими, биологическими факторами или дефицитом субстратов метаболизма;
- 2) перегрузочная, которая развивается на фоне повышенной работы миокарда по преодолению избыточного давления на путях изгнания крови из камер сердца (например, при гипертонической болезни); в связи с перегрузкой сердца увеличенным объемом кро-
ви (например, при наличии внутрисердечных шунтов) или при сочетании этих двух факторов (перегрузка объемом и давлением).
Очень часто течение миокардиальной сердечной недостаточности усугубляется присоединением ее перегрузочной формы.
По преимущественному поражению камер сердца сердечная недостаточность подразделяется на левожелудочковую, правожелудочковую и тотальную, каждая из которых, в свою очередь, может быть по характеру течения острой и хронической.
Левожелудочковая недостаточность встречается значительно чаще правожелудочковой, несмотря на то что левый желудочек более приспособлен к повышенным нагрузкам. Это связано с тем, что различные патологические состояния чаще приводят к перегрузке левого желудочка. При острой левожелудочковой недостаточности такими состояниями могут быть артериальная гипертензия (прежде всего гипертонический криз), инфаркт миокарда и обратимая ишемия левого желудочка, отрыв папиллярной мышцы с пролапсом митрального клапана и др. Острая левожелудочковая недостаточность клинически проявляется в виде сердечной астмы или отека легких. С морфологической точки зрения сердечная астма соответствует интерстициальному отеку, а отек легких — альвеолярному, или истинному, отеку легких.
Хроническая левожелудочковая недостаточность — это медленно формирующееся патологическое состояние, при котором нагрузка на левый желудочек превышает его способность совершать работу. Следует отметить, что этиологические факторы острой и хронической недостаточности сердца существенно различаются. Хроническая левожелудочковая недостаточность осложняет течение только хронических заболеваний сердца и сосудов.
Правожелудочковая недостаточность характеризуется развитием застойных явлений в большом круге кровообращения. При этом увеличивается кровенаполнение печени и соответственно ее размеры, нарушается экскреторная функция почек, происходит задержка воды в организме и появляются периферические отеки. Различают острую и хроническую правожелудочковую недостаточность. Наиболее частой причиной острой правожелудочковой недостаточности является распространение крупноочагового инфаркта левого желудочка на правые отделы сердца, реже — изолированный некроз миокарда правого желудочка. Очень часто причиной правожелудочковой недостаточности является легочная гипертензия, которая также может развиваться остро и хронически (см. раздел 15.2.2).
К весьма распространенным проявлениям перегрузки правого желудочка относятся легочное сердце и эмболия легочной артерии.
Понятие «легочное сердце» включает в себя легочную гипертензию, гипертрофию правого желудочка, его дилатацию и сердечную недостаточность. Однако в ряде случаев легочное сердце может проявляться только некоторыми из вышеназванных признаков. Так, длительное повышение артериального давления в легочных сосудах обычно приводит к гипертрофии правого желудочка с последующим развитием сердечной недостаточности по большому кругу. Если легочная гипертензия быстро прогрессирует, то гипертрофия правого желудочка не успевает развиться и дилатация непосредственно переходит в правожелудочковую недостаточность. Такая ситуация может иметь место и при тромбоэмболии легочной артерии, пневмотораксе, астматическом статусе и распространенной пневмонии, когда артериальное давление в малом круге повышается в течение нескольких суток или даже часов.
Синдром эмболии легочной артерии — это патологическое состояние, которое развивается при попадании эмболов в русло легочной артерии и характеризуется болями в грудной клетке, одышкой, цианозом с одновременным появлением тахикардии и признаков коллапса. Эмболия легочной артерии особенно опасна тем, что может вызвать рефлекторную остановку сердца и внезапную смерть больного. Если же пациент не погиб в первые минуты и часы, то спустя сутки-двое после эмболии формируется инфаркт легкого и так называемая инфаркт-пневмония, которая может послужить причиной развития легочного сердца.
Наиболее часто эмболия легочной артерии вызывается тромбами, мигрирующими из венозной системы и правых отделов сердца или из «левого сердца» при наличии дефектов межжелудочковой и межпредсердной перегородок. Наряду с этим причиной эмболии артерий малого круга кровообращения могут стать пузырьки воздуха или капли жира, попавшие в венозную систему при нарушении процедуры внутривенной инфузии или при травмах трубчатых костей.
При закупорке мелких ветвей легочной артерии выраженные клинические симптомы могут отсутствовать. Именно поэтому тромбоэмболия легочной артерии диагностируется только у 30% больных. Однако на вскрытии погибших пациентов, вместо ожидавшейся полной закупорки легочной артерии, иногда обнаруживали тромб диаметром всего 3−4 мм в одной из ветвей легочной артерии (крупные сосуды), не закрывавший ни одну из них даже на треть. Этот факт подчеркивает важную роль нарушений нейрогуморальной регуляции гемодинамики в патогенезе легочной гипертензии.
По течению хроническая сердечная недостаточность делится на три стадии. Первая стадия — начальная. На этом этапе общие симптомы хронической сердечной недостаточности (одышка, тахикардия) появляются только во время физической нагрузки. Местные симптомы (цианоз) обычно отсутствуют. Во второй стадии появляются не только общие, но и местные симптомы. Помимо одышки и тахикардии при физической нагрузке появляются цианоз, признаки декомпенсации функционального состояния внутренних органов, асцит и анасарка. Третья стадия — конечная, дистрофическая. В этот период развивается тотальная застойная недостаточность, затрагивающая большой и малый круг кровообращения. Формируется почечная, печеночная и легочная недостаточность. Третья стадия является терминальной стадией хронической сердечной недостаточности.
Механизмы компенсации гемодинамики при сердечной недостаточности Здоровый организм обладает многообразными механизмами, обеспечивающими своевременную разгрузку сосудистого русла от избытка жидкости. При сердечной недостаточности «включаются» компенсаторные механизмы, направленные на сохранение нормальной гемодинамики. Эти механизмы в условиях острой и хронической недостаточности кровообращения имеют много общего, вместе с тем между ними отмечаются существенные различия.
Как и при острой, так и при хронической сердечной недостаточности все эндогенные механизмы компенсации гемодинамических нарушений можно подразделить на интракардиальные: компенсаторная гиперфункция сердца (механизм Франка-Старлинга, гомеометрическая гиперфункция), гипертрофия миокарда и экстракардиальные: разгрузочные рефлексы Бейнбриджа, Парина, Китаева, активация выделительной функции почек, депонирование крови в печени и селезенке, потоотделение, испарение воды со стенок легочных альвеол, активация эритропоэза и др. Такое деление в некоторой степени условно, поскольку реализация как интра-, так и экстракардиальных механизмов находится под контролем нейрогуморальных регуляторных систем.
Механизмы компенсации гемодинамических нарушений при острой сердечной недостаточности. На начальной стадии систолической дисфункции желудочков сердца включаются интракардиальные факторы компенсации сердечной недостаточности, важнейшим из которых является механизм Франка-Старлинга (гетерометрический механизм компенсации, гетерометрическая гиперфункция сердца). Реализацию его можно представить следующим образом. Нарушение сократительной функции сердца влечет за собой уменьшение ударного объема крови и гипоперфузию почек. Это способствует активации РААС, вызывающей задержку воды в организме и увеличение объема циркулирующей крови. В условиях возникшей гиперволемии происходит усиленный приток венозной крови к сердцу, увеличение диастолического кровенаполнения желудочков, растяжение миофибрилл миокарда и компенсаторное повышение силы сокращения сердечной мышцы, которое обеспечивает прирост ударного объема. Однако если конечное диастолическое давление повышается более чем на 18−22 мм рт.ст., возникает чрезмерное перерастяжение миофибрилл. В этом случае компенсаторный механизм Франка-Старлинга перестает действовать, а дальнейшее увеличение конечного диастолического объема или давления вызывает уже не подъем, а снижение ударного объема.
Наряду с внутрисердечными механизмами компенсации при острой левожелудочковой недостаточности запускаются разгрузочные экстракардиальные рефлексы, способствующие возникновению тахикардии и увеличению минутного объема крови (МОК). Одним из наиболее важных сердечно-сосудистых рефлексов, обеспечивающих увеличение МОК, является рефлекс Бейнбриджа увеличение частоты сердечных сокращений в ответ на увеличение объема циркулирующей крови. Этот рефлекс реализуется при раздражении механорецепторов, локализованных в устье полых и легочных вен. Их раздражение передается на центральные симпатические ядра продолговатого мозга, в результате чего происходит повышение тонической активности симпатического звена вегетативной нервной системы, и развивается рефлекторная тахикардия. Рефлекс Бейнбриджа направлен на увеличение минутного объема крови.
Рефлекс Бецольда-Яриша — это рефлекторное расширение артериол большого круга кровообращения в ответ на разражение механои хеморецепторов, локализованных в желудочках и предсердиях.
В результате возникает гипотония, которая сопровождается брадикардией и временной остановкой дыхания. В реализации этого рефлекса принимают участие афферентные и эфферентные волокна n. vagus. Этот рефлекс направлен на разгрузку левого желудочка.
К числу компенсаторных механизмов при острой сердечной недостаточности относится и повышение активности симпатоадреналовой системы, одним из звеньев которого является высвобождение норадреналина из окончаний симпатических нервов, иннервирующих сердце и почки. Наблюдаемое при этом возбуждение в-адренорецепторов миокарда ведет к развитию тахикардии, а стимуляция подобных рецепторов в клетках ЮГА вызывает усиленную секрецию ренина. Другим стимулом секреции ренина является снижение почечного кровотока в результате вызванной катехоламинами констрикции артериол почечных клубочков. Компенсаторное по своей природе усиление адренергического влияния на миокард в условиях острой сердечной недостаточности направлено на увеличение ударного и минутного объемов крови. Положительный инотропный эффект оказывает также ангиотензин-II. Однако эти компенсаторные механизмы могут усугубить сердечную недостаточность, если повышенная активность адренергической системы и РААС сохраняется достаточно продолжительное время (более 24 ч).
Все сказанное о механизмах компенсации сердечной деятельности в одинаковой степени относится как к лево-, так и к правожелудочковой недостаточности. Исключением является рефлекс Парина, действие которого реализуется только при перегрузке правого желудочка, наблюдаемой при эмболии легочной артерии.
Рефлекс Ларина — это падение артериального давления, вызванное расширением артерий большого круга кровообращения, снижением минутного объема крови в результате возникающей брадикардии и уменьшением объема циркулирующей крови из-за депонирования крови в печени и селезенке. Кроме того, для рефлекса Парина характерно появление одышки, связанной с наступающей гипоксией мозга. Полагают, что рефлекс Парина реализуется за счет усиления тонического влияния n.vagus на сердечно-сосудистую систему при эмболии легочных артерий.
Механизмы компенсации гемодинамических нарушений при хронической сердечной недостаточности. Основным звеном патогенеза хронической сердечной недостаточности является, как известно, постепенно нарастающее снижение сократительной функции ми;
окарда и падение сердечного выброса. Происходящее при этом уменьшение притока крови к органам и тканям вызывает гипоксию последних, которая первоначально может компенсироваться усиленной тканевой утилизацией кислорода, стимуляцией эритропоэза и т. д. Однако этого оказывается недостаточно для нормального кислородного обеспечения органов и тканей, и нарастающая гипоксия становится пусковым механизмом компенсаторных изменений гемодинамики.
Интракардиальные механизмы компенсации функции сердца. К ним относятся компенсаторная гиперфункция и гипертрофия сердца. Эти механизмы являются неотъемлемыми компонентами большинства приспособительных реакций сердечно-сосудистой системы здорового организма, но в условиях патологии могут превратиться в звено патогенеза хронической сердечной недостаточности.
Компенсаторная гиперфункция сердца выступает как важный фактор компенсации при пороках сердца, артериальной гипертензии, анемии, гипертонии малого круга и других заболеваниях. В отличие от физиологической гиперфункции она является длительной и, что существенно, непрерывной. Несмотря на непрерывность, компенсаторная гиперфункция сердца может сохраняться в течение многих лет без явных признаков декомпенсации насосной функции сердца.
Увеличение внешней работы сердца, связанное с подъемом давления в аорте (гомеометрическая гиперфункция), приводит к более выраженному возрастанию потребности миокарда в кислороде, чем перегрузка миокарда, вызванная повышением объема циркулирующей крови (гетерометрическая гиперфункция). Иными словами, для осуществления работы в условиях нагрузки давлением мышца сердца использует гораздо больше энергии, чем для выполнения той же работы, связанной с нагрузкой объемом, а следовательно, при стойкой артериальной гипертензии гипертрофия сердца развивается быстрее, чем при увеличении объема циркулирующей крови. Например, при физической работе, высотной гипоксии, всех видах клапанной недостаточности, артериовенозных фистулах, анемии гиперфункция миокарда обеспечивается за счет увеличения минутного объема сердца. При этом систолическое напряжение миокарда и давление в желудочках возрастают незначительно, и гипертрофия развивается медленно. В то же время при гипертонической болезни, гипертензии малого круга, стенозах клапанных отверстий развитие гиперфункции связано с повышением напряжения миокарда при незначительно измененной амплитуде сокращений. В этом случае гипертрофия прогрессирует достаточно быстро.
Гипертрофия миокарда - это увеличение массы сердца за счет увеличения размеров кардиомиоцитов. Существуют три стадии компенсаторной гипертрофии сердца.
Первая, аварийная, стадия характеризуется, прежде всего, увеличением интенсивности функционирования структур миокарда и, по сути, представляет собой компенсаторную гиперфункцию еще не гипертрофированного сердца. Интенсивность функционирования структур — это механическая работа, приходящаяся на единицу массы миокарда. Увеличение интенсивности функционирования структур закономерно влечет за собой одновременную активацию энергообразования, синтеза нуклеиновых кислот и белка. Указанная активация синтеза белка происходит таким образом, что вначале увеличивается масса энергообразующих структур (митохондрий), а затем — масса функционирующих структур (миофибрилл). В целом увеличение массы миокарда приводит к тому, что интенсивность функционирования структур постепенно возвращается к нормальному уровню.
Вторая стадия — стадия завершившейся гипертрофии — характеризуется нормальной интенсивностью функционирования структур миокарда и соответственно нормальным уровнем энергообразования и синтеза нуклеиновых кислот и белков в ткани сердечной мышцы. При этом потребление кислорода на единицу массы миокарда остается в границах нормы, а потребление кислорода сердечной мышцей в целом увеличено пропорционально возрастанию массы сердца. Увеличение массы миокарда в условиях хронической сердечной недостаточности происходит за счет активации синтеза нуклеиновых кислот и белков. Пусковой механизм этой активации изучен недостаточно. Считается, что определяющую роль здесь играет усиление трофического влияния симпатоадреналовой системы. Эта стадия процесса совпадает с длительным периодом клинической компенсации. Содержание АТФ и гликогена в кардиомиоцитах также находится при этом в пределах нормы. Подобные обстоятельства придают относительную устойчивость гиперфункции, но вместе с тем не предотвращают исподволь развивающихся в данной стадии нарушений обмена и структуры миокарда. Наиболее ранними признаками таких нарушений являются значительное увеличение концентрации лактата в миокарде, а также умеренно выраженный кардиосклероз.
Третья стадия прогрессирующего кардиосклероза и декомпенсации характеризуется нарушением синтеза белков и нуклеиновых кислот в миокарде. В результате нарушения синтеза РНК, ДНК и белка в кардиомиоцитах наблюдается относительное уменьшение массы митохондрий, что ведет к торможению синтеза АТФ на единицу массы ткани, снижению насосной функции сердца и прогрессированию хронической сердечной недостаточности. Ситуация усугубляется развитием дистрофических и склеротических процессов, что способствует появлению признаков декомпенсации и тотальной сердечной недостаточности, завершающейся гибелью пациента. Компенсаторная гиперфункция, гипертрофия и последующая декомпенсация сердца — это звенья единого процесса.
Механизм декомпенсации гипертрофированного миокарда включает следующие звенья:
- 1. Процесс гипертрофии не распространяется на коронарные сосуды, поэтому число капилляров на единицу объема миокарда в гипертрофированном сердце уменьшается (рис. 15−11). Следовательно, кровоснабжение гипертрофированной сердечной мышцы оказывается недостаточным для выполнения механической работы.
- 2. Вследствие увеличения объема гипертрофированных мышечных волокон уменьшается удельная поверхность клеток, в связи с этим ухудшаются условия для поступления в клетки питательных веществ и выделения из кардиомиоцитов продуктов метаболизма.
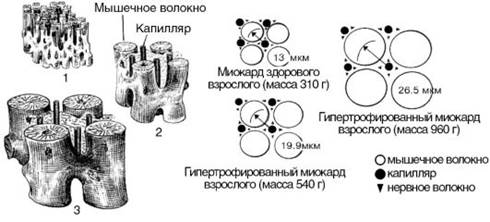
Рис. 5−11. Гипертрофия миокарда: 1 — миокард здорового взрослого; 2 — гипертрофированный миокард взрослого (масса 540 г); 3 — гипертрофированный миокард взрослого (масса 960 г).
- 3. В гипертрофированном сердце нарушается соотношение между объемами внутриклеточных структур. Так, увеличение массы митохондрий и саркоплазматического ретикулума (СПР) отстает от увеличения размеров миофибрилл, что способствует ухудшению энергоснабжения кардиомиоцитов и сопровождается нарушением аккумуляции Са2+ в СПР. Возникает Са2±перегрузка кардиомиоцитов, что обеспечивает формирование контрактуры сердца и способствует уменьшению ударного объема. Кроме того, Са2±перегрузка клеток миокарда повышает вероятность возникновения аритмий.
- 4. Проводящая система сердца и вегетативные нервные волокна, иннервирующие миокард, не подвергаются гипертрофии, что также способствует возникновению дисфункции гипертрофированного сердца.
- 5. Активируется апоптоз отдельных кардиомиоцитов, что способствует постепенному замещению мышечных волокон соединительной тканью (кардиосклероз).
В конечном итоге гипертрофия утрачивает приспособительное значение и перестает быть полезной для организма. Ослабление сократительной способности гипертрофированного сердца происходит тем скорее, чем сильнее выражены гипертрофия и морфологические изменения в миокарде.
Экстракардиальные механизмы компенсации функции сердца. В отличие от острой сердечной недостаточности роль рефлекторных механизмов экстренной регуляции насосной функции сердца при хронической сердечной недостаточности сравнительно невелика, поскольку нарушения гемодинамики развиваются постепенно на протяжении нескольких лет. Более или менее определенно можно говорить о рефлексе Бейнбриджа, который «включается» уже на стадии достаточно выраженной гиперволемии.
Особое место среди «разгрузочных» экстракардиальных рефлексов занимает рефлекс Китаева, который «запускается» при митральном стенозе. Дело в том, что в большинстве случаев проявления правожелудочковой недостаточности связаны с застойными явлениями в большом круге кровообращения, а левожелудочковой — в малом. Исключение составляет стеноз митрального клапана, при котором застойные явления в легочных сосудах вызваны не декомпенсацией левого желудочка, а препятствием току крови через левое атриовентрикулярное отверстие — так называемым «первым (анатомическим) барьером». При этом застой крови в легких способствует развитию правожелудочковой недостаточности, в генезе которой рефлекс Китаева играет важную роль.
Рефлекс Китаева — это рефлекторный спазм легочных артериол в ответ на повышение давления в левом предсердии. В результате возникает «второй (функциональный) барьер», который первоначально играет защитную роль, предохраняя легочные капилляры от чрезмерного переполнения кровью. Однако затем этот рефлекс приводит к выраженному повышению давления в легочной артерии — развивается острая легочная гипертензия. Афферентное звено этого рефлекса представлено n. vagus, a эфферентное — симпатическим звеном вегетативной нервной системы. Негативной стороной данной приспособительной реакции является подъем давления в легочной артерии, приводящий к увеличению нагрузки на правое сердце.
Однако ведущую роль в генезе долговременной компенсации и декомпенсации нарушенной сердечной функции играют не рефлекторные, а нейрогуморальные механизмы, важнейшим из которых является активация симпатоадреналовой системы и РААС. Говоря об активации симпатоадреналовой системы у пациентов с хронической сердечной недостаточностью, нельзя не указать, что у большинства из них уровень катехоламинов в крови и моче находится в пределах нормы. Этим хроническая сердечная недостаточность отличается от острой сердечной недостаточности.
Механизмы декомпенсации сердечной недостаточности Параллельно с интраи экстракардиальными компенсаторными изменениями, которые развиваются при сердечной недостаточности, появляются и постепенно прогрессируют повреждения сердечной мышцы, приводящие к снижению ее сократительной способности. На определенной стадии процесса такие явления могут быть обратимыми. При продолжении или усилении действия причинного фактора, вызвавшего сердечную недостаточность, а также при срыве механизмов компенсации развиваются необратимые диффузные изменения миокарда с характерной клинической картиной декомпенсированной сердечной недостаточности.
Патогенез сердечной недостаточности представляется следующим образом. Многочисленный ряд примеров патологии сер;
дечной деятельности (кардиомиопатии, нарушения коронарной перфузии и др.) индуцирует кислородное голодание миокарда. Известно, что в условиях нормального кровоснабжения важным энергетическим субстратом для сердечной мышцы являются свободные жирные кислоты, глюкоза и молочная кислота. Гипоксия приводит к нарушению процессов аэробного окисления субстратов в цикле Кребса, к угнетению окисления НАДН в дыхательной цепи митохондрий. Все это способствует накоплению недоокисленных продуктов метаболизма свободных жирных кислот и глюкозы (ацил-КоА, лактат). Усиленное образование ацил-КоА в кардиомиоцитах негативно сказывается на энергетическом метаболизме клетки. Дело в том, что ацил-КоА является ингибитором аденилаттранслоказы — фермента, который осуществляет транспорт АТФ из митохондрий в саркоплазму. Аккумуляция ацил-КоА приводит к нарушению этого транспорта, усугубляя энергетический дефицит в клетке.
Единственным источником энергии для кардиомиоцитов становится анаэробный гликолиз, интенсивность которого в условиях гипоксии резко возрастает. Однако «коэффициент полезного действия» анаэробного гликолиза по сравнению с эффективностью энергопродукции в цикле Кребса намного ниже. В силу этого анаэробный гликолиз не в состоянии полностью возместить энергетические потребности клетки. Так, при анаэробном расщеплении одной молекулы глюкозы образуются всего две молекулы АТФ, в то время как при окислении глюкозы до углекислого газа и воды — 32 молекулы АТФ. Нехватка высокоэнергетических фосфатов (АТФ и креатинфосфата) приводит к нарушению энергозависимого процесса удаления ионов кальция из саркоплазмы кардиомиоцитов и возникновению кальциевой перегрузки миокарда.
В норме увеличение концентрации Ca2+ в кардиомиоцитах вызывает образование мостиков между цепочками актина и миозина, что является основой сокращения клеток. Вслед за этим происходит удаление избытка ионов кальция из саркоплазмы и развитие диастолы. Кальциевая перегрузка клеток миокарда при его ишемии ведет к остановке процесса сокращения — расслабления в стадии систолы, формируется контрактура миокарда — состояние, при котором кардиомиоциты перестают расслабляться. Возникшая зона асистолии характеризуется повышенным тканевым напряжением, что ведет к сдавлению коронарных сосудов и связанному с этим усугублению дефицита коронарного кровотока.
Ионы Са активируют фосфолипазу А2, которая катализирует расщепление фосфолипидов. В результате этого образуются одна молекула свободной жирной кислоты и одна молекула лизофосфатида. Свободные жирные кислоты обладают детергентоподобным действием и в случае избыточного их накопления в миокарде могут повреждать мембраны кардиомиоцитов. Еще более выраженный кардиотоксический эффект оказывают лизофосфатиды. Особенно токсичен лизофосфатидилхолин, который может провоцировать аритмии. В настоящее время роль свободных жирных кислот и лизофосфатидов в патогенезе ишемического повреждения сердца никем не оспаривается, однако молекулярная природа необратимого повреждения кардиомиоцитов не сводится только к накоплению этих веществ в клетках сердечной мышцы. Кардиотоксическими свойствами могут обладать и другие продукты метаболизма, например активные формы кислорода (АФК).
К АФК относятся супероксидный радикал (O2*-) и гидроксильный радикал O2*-, которые обладают высокой окислительной активностью. Источником АФК в кардиомиоцитах является дыхательная цепь митохондрий и прежде всего цитохромы, которые в условиях гипоксии переходят в восстановленное состояние и могут быть донорами электронов, «передавая» их молекулам кислорода с образованием не молекулы воды, как это происходит в норме, а супероксидного радикала (O2*-). Кроме того, образование свободных радикалов катализируется ионами металлов с переменной валентностью (прежде всего ионами железа), которые всегда присутствуют в клетке. АФК взаимодействуют с молекулами белков и полиненасыщенных жирных кислот, превращая их в свободные радикалы. Вновь образованные радикалы могут, в свою очередь, взаимодействовать с другими молекулами белков и жирных кислот, индуцируя дальнейшее образование свободных радикалов. Таким образом, реакция может принимать цепной и разветвленный характер. Если пероксидации подвергаются белки ионных каналов, то происходит нарушение процессов ионного транспорта. Если гидроперекиси образуются из молекул ферментов, последние теряют свою каталитическую активность.
Образование гидроперекисей полиненасыщенных жирных кислот, входящих в молекулярную структуру мембранных фосфолипидов, способствует изменению биологических свойств мембран. В отличие от жирных кислот гидроперекиси являются водорастворимыми веществами, и появление их в структуре гидрофобного фосфолипидного матрикса клеточных мембран приводит к формированию пор, пропускающих ионы и молекулы воды. Кроме того, изменяется активность мембраносвязанных ферментов.
Процесс возникновения гидроперекисей жирных кислот является одним из звеньев перекисного окисления липидов (ПОЛ), которое включает в себя свободнорадикальное образование альдегидов и кетонов — продуктов ПОЛ. Согласно концепции Ф. З. Меерсона, продукты ПОЛ обладают кардиотоксическими свойствами, их накопление в клетке приводит к повреждению сарколеммы, а также лизосомальных и митохондриальных мембран. На заключительном этапе повреждения, предшествующем гибели клеток, особая роль отводится активации протеолитических ферментов. Обычно эти энзимы находятся в цитоплазме кардиомиоцитов в неактивном состоянии или локализованы внутри лизосом, мембраны которых изолируют их от структурных элементов клетки. В связи с этим в норме протеазы не оказывают цитотоксического действия. В условиях ишемии перегрузка кардиомиоцитов ионами кальция и закисление цитоплазмы за счет накопления лактата приводят к активации внутриклеточных протеаз. Кроме того, повышение проницаемости лизосомальных мембран под действием фосфолипаз и продуктов ПОЛ способствует выходу активных протеолитических ферментов в саркоплазму. Конечным звеном этой патогенетической цепочки является некроз кардиомиоцитов в зоне ишемии и их аутолиз.
Важно отметить, что первыми погибают только те кардиомиоциты, которые отличаются высокой интенсивностью энергетического метаболизма и соответственно повышенной потребностью в кислороде. В то же время фибробласты и клетки проводящей системы менее зависимы от доставки кислорода и сохраняют свою жизнеспособность. Функциональная активность фибробластов обеспечивает процессы рубцевания.
Клетки проводящей системы, сохраняя жизнеспособность в условиях кислородного голодания, существенно изменяют свои электрофизиологические характеристики, что может способствовать возникновению аритмий. В результате повреждения мембран и снижения образования АТФ изменяется активность К+/ Na+-АТФазы, что сопровождается усиленным поступлением натрия в кардиомиоциты и выходом из них калия. Это увеличивает электрическую нестабильность миокарда и способствует развитию аритмий.
Гипоксическая сократительная дисфункция сердца усугубляется нарушением процессов нейрогуморальной регуляции функционального состояния миокарда. Сердечные боли, приступы аритмии и другие нарушения являются для организма стрессором, т. е. воздействием чрезмерной силы, на которое организм, как и на любое стрессорное воздействие, реагирует активацией симпатоадреналовой системы. При этом происходит выброс катехоламинов из надпочечников и симпатических нервных терминалей. Однако, как и любой другой компенсаторный процесс, активация симпатоадреналовой системы в конце концов приобретает негативную окраску. Наступает период декомпенсации. Схематично последовательность событий представлена на рисунке 15−12.
В настоящее время установлено, что при хронической активации симпатоадреналовой системы происходят постепенная Са2± перегрузка кардиомиоцитов и их контрактура, нарушается целостность сарколеммы. При гиперактивации адренергической системы формируется электрическая нестабильность миокарда. Последняя способствует возникновению фибрилляции желудочков сердца, поэтому каждый третий пациент при хронической сердечной недостаточности погибает внезапно, иногда сердечная смерть наступает на фоне внешнего благополучия и положительной клинической динамики.
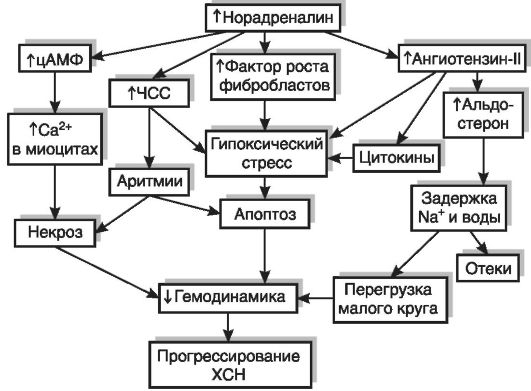
Рис. 15−12. Роль симпатоадреналовой и ренин-ангиотензин-альдостероновой систем в патогенезе хронической сердечной недостаточности: ХСН — хроническая сердечная недостаточность; ЧСС — частота сердечных сокращений Адренергическая тахикардия сопровождается повышением потребности миокарда в кислороде, что наряду с Са2±перегрузкой еще больше усугубляет энергетический дефицит в клетках миокарда. Включается защитно-приспособительный механизм, получивший название гибернации (спячки) кардиомиоцитов. Часть клеток перестает сокращаться и отвечать на внешние стимулы, потребляя при этом минимум энергии и экономя кислород для активно сокращающихся кардиомиоцитов. Таким образом, количество обеспечивающих насосную функцию сердца клеток миокарда может существенно уменьшиться, способствуя усугублению сердечной недостаточности.
Кроме того, гиперактивация симпатоадреналовой системы усиливает секрецию ренина почками, выступая в роли стимулятора РААС. Образующийся ангиотензин-II оказывает ряд негативных эффектов на сердечно-сосудистую систему. Он способствует увеличению адренореактивности сердца и сосудов, усиливая тем самым кардиотоксическое действие катехоламинов. Одновременно этот пептид увеличивает периферическое сопротивление кровеносных сосудов, что, безусловно, способствует увеличению постнагрузки на сердце и весьма негативно сказывается на гемодинамике. Кроме того, ангиотензин-II может самостоятельно или через активацию образования цитокинов (биологически активные вещества белковой природы, образующиеся в миокарде и других тканях) стимулировать программируемую гибель кардиомиоцитов («апоптоз»).
Наряду с отмеченным, повышение уровня ангиотензина-II негативно сказывается на состоянии водно-солевого гомеостаза, поскольку этот пептид активирует секрецию альдостерона. В результате в организме задерживается избыточное количество воды и натрия. Задержка натрия повышает осмолярность крови, в ответ на которую происходит активация секреции антидиуретического гормона, что приводит к уменьшению диуреза и еще большей гидратации организма. В итоге повышается объем циркулирующей крови и увеличивается преднагрузка на сердце. Гиперволемия ведет к раздражению механорецепторов, локализованных в устье полых и легочных вен, «включается» рефлекс Бейнбриджа, возникает рефлекторная тахикардия, что еще больше увеличивает нагрузку на миокард и потребность сердечной мышцы в кислороде.
Создается «порочный круг», разорвать который можно только с помощью определенных фармакологических воздействий. Ко всему этому присоединяется повышение гидростатического давления в микрососудистом русле, что способствует выходу жидкой части крови в ткани и формированию отеков. Последние сдавливают ткани, что усугубляет нарушение микроциркуляции и еще больше усиливает тканевую гипоксию. При дальнейшем прогрессировании недостаточности кровообращения нарушаются и другие виды обмена, в том числе и белковый, что приводит к дистрофическим изменениям в органах и тканях, нарушению их функции. В конечной стадии хронической сердечной недостаточности развиваются кахексия, маскируемая отеками, гипопротеинемия, появляются признаки почечной и печеночной декомпенсации.