Нарушение синтеза белка

Наиболее частая форма классической ФКУ проявляется характерным симптомокомплексом: умственная отсталость, судорожный синдром, склонность к экземе, нарушение пигментного обмена. От больных исходит специфический «мышиный» запах, обусловленный высоким содержанием фенольных кислот в биологических жидкостях. Эти симптомы появляются в первый год жизни, и больные при отсутствии лечения, как правило… Читать ещё >
Нарушение синтеза белка (реферат, курсовая, диплом, контрольная)
I. Нарушения синтеза белка приобретенного характера обусловлены преимущественно следующими причинами.
- § Различные виды алиментарной недостаточности (полное, неполное голодание, отсутствие в пище незаменимых аминокислот, нарушение определенного количественного соотношения между незаменимыми аминокислотами, поступающими в организм). Отсутствие в клетках хотя бы одной незаменимой аминокислоты прекращает синтез белка в целом.
- § Дефицит энергии при гипоксии.
- § Дефицит анаболических гормонов.
- § Избыток катаболических гормонов и факторов (ФНО при лихорадке и опухолях).
- § Опухоли.
- § Дефицит витаминов.
- § Нарушение иннервации вызывает дефицит трофогенов и развитие трофических язв.
- § Нарушение синтеза белка вызывают некоторые антибиотики. Так, «ошибки» в считывании генетического кода могут возникнуть под влиянием стрептомицина, неомицина и других антибиотиков. Тетрациклины тормозят присоединение новых аминокислот к растущей полипептидной цепи (образование прочных ковалентных связей между ее цепями), препятствуя расщеплению нитей ДНК.
Изменение активности ферментных систем клеток, участвующих в синтезе белка, носят в основном наследственный характер
- § Гемоглобинопатии — аномалии, связанные с нарушением механизма синтеза белкового компонента гемоглобина при нормальной структуре гема. Выявлено более 15 видов аномальных молекул гемоглобина, где в альфаили бета-цепи произошла замена одной из АК. Известно несколько видов мутантных гемоглобинов, где произошла замена остатка гистидина, который связывает железо гема с белковой частью молекулы, на другие АК. Получаются так называемые М-гемоглобины, которые не способны транспортировать кислород.
- § Глютеновая энтеропатия (целиакия) характеризуется нарушением всасывания вследствие иммуноопосредованного воспалительного повреждения слизистой оболочки тонкой кишки, пусковым фактором которого является потребление в пищу пшеничного глютена или родственных белков ржи и ячменя у генетически предрасположенных людей. Повреждающее действие на слизистую оболочку тонкой кишки оказывает глиадин — один из основных компонентов растительного белка глютена. Глиадин связывается со специфическим рецептором энтероцитов, взаимодействует с Т-лимфоцитами. Образующиеся лимфокины и антитела повреждают энтероциты ворсинок. На повреждающее действие глиадина слизистая оболочка отвечает атрофией и инфильтрацией иммунокомпетентными клетками.
Ранее считалось, что целиакия — болезнь генетической природы, при которой имеется дефицит ферментов, расщепляющих один из компонентов белка клейковины злаков до аминокислот, из-за чего в организме накапливаются продукты его неполного гидролиза.
- § Фенилкетонурия.
- § Альбинизм.
- § Алкаптонурия.
Наследственные энзимопатии, связанные с обменом аминокислот. Фармакологическая коррекция разных типов фенилкетонурии Таблица 16.1.
Наследственные энзимопатии, связанные с обменом аминокислот.
Аминокислота. | Превращения в норме. | Ферментный дефицит. | Клинические проявления. | Прочее. |
Фенилаланин. | Тирозин. | фенилаланингидроксилаза. |
| Диагностика: 5% трихлоруксусное железо > оливково-зеленое окрашивание мочи. |
Тирозин. | Меланин Тироксин. | Тирозиназа, фермент йодирования. |
| |
Триптофан. | Серотонин. |
|
![Патогенез основных видов наследственных энзимопатий, связанных с обменом аминокислот [по А.Ш. Зайчику, Л.П. Чурилову, 2000].](/img/s/9/67/2206567_1.png)
Рис. 16.8. Патогенез основных видов наследственных энзимопатий, связанных с обменом аминокислот [по А. Ш. Зайчику, Л. П. Чурилову, 2000].
Алкаптонурия — аутосомно-рецессивная болезнь, на примере которой сэр Арчибальд Гаррод (1909) создал концепцию метаболического блока. Причина заболевания — дефект оксидазы промежуточного продукта распада фенилаланина и тирозина — гомогентизиновой кислоты.
В норме фермент n-оксифенилпируватдезоксигеназа в присутствии витамина С, превращает полученный из тирозина n-оксифенилнируват в гомогентизиновую кислоту. Затем, гомогентизиновая кислота должна в почках окислиться в присутствии Fe+2 и глутатиона до 4-малеилацетоуксусной кислоты. Если этот процесс тормозится, то накопление гомогентизиновой кислоты ведет к ее превращению полифенолоксидазой в хиноновые полифенолы, составляющие так называемый «охронозный пигмент», выводимый почками. Это обусловливает потемнение мочи больных на воздухе.
Гомогентизиновая кислота ингибирует фермент лизилгидроксилазу, участвующий в синтезе коллагена, а охронозный пигмент (алкаптон) не полностью экскретируется с мочой, откладывается в основном веществе хрящей и других соединительно-тканных образований и делает их хрупкими, что со временем (после 30-ти лет) вызывает кальцификацию и дегенеративный артрит позвоночника, а также крупных суставов конечностей. Первыми проявлениями болезни могут быть пигментация склер и хрящей ушных раковин.
Радикально болезнь не лечится. Степень остеохондропатии можно уменьшить, защищая активность лизилгидроксилазы большими дозами аскорбиновой кислоты.
Фенилаланин — незаменимая аминокислота. Помимо включения в процесс биосинтеза белков, фенилаланин в печени метаболизируется двумя путями. В норме большая часть фенилаланина превращается в тирозин. Реакция катализируется фенилаланингидроксилазой при участии дигидробиоптерина.
Известны две гетерогенные группы наследственных нарушений обмена фенилаланина, сопровождающихся фенилаланинемией:
- 1) обусловленные дефектом фенилаланингидроксилазы,
- 2) являющиеся следствием дефектов ферментов, участвующих в синтезе и метаболизме тетрагидробиоптерина.
Фенилкетонурия (ФКУ) — наследственное заболевание, обусловленное мутациями в гене фенилаланингидроксилазы (классическая ФКУ).
Наиболее частая форма классической ФКУ проявляется характерным симптомокомплексом: умственная отсталость, судорожный синдром, склонность к экземе, нарушение пигментного обмена. От больных исходит специфический «мышиный» запах, обусловленный высоким содержанием фенольных кислот в биологических жидкостях. Эти симптомы появляются в первый год жизни, и больные при отсутствии лечения, как правило, не доживают до 30 лет.
В патогенезе симптомов ФКУ основную роль играют нарушения обмена циклических аминокислот:
- * увеличение концентрации фенилаланина ограничивает транспорт тирозина и триптофана через гематоэнцефалический барьер (ГЭБ) и тормозит синтез нейромедиаторов;
- * накопление фенилаланина в клетках мозга нарушает реакции синтеза цереброзидсульфатов, участвующих в защите мозга от демиелинизации;
- * фенилаланин и его производные фенольные кислоты оказывают нейротоксическое действие. Они являются ингибиторами тирозингидроксилазы и триптофангидроксилазы — ключевых ферментов синтеза нейромедиаторов: дофамина, норадреналина, серотонина.
Для предотвращения токсического действия фенилаланина и фенилпирувата белковая пища просто заменяется набором аминокислот без фенилаланина.
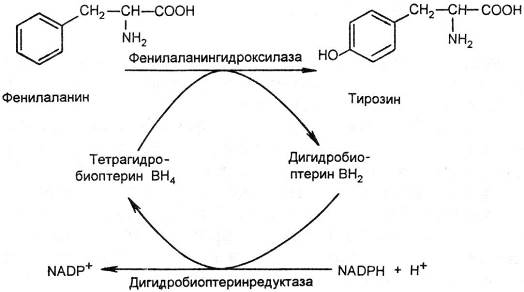
Рис. 16.9. Превращение фенилаланина в тирозин [по Е. С. Северину, 2000]. ВН4 синтезируется в организме из ГТФ, но в недостаточном количестве. Его регенерация при участии дигидробиоптеринредуктазы является необходимым звеном в превращении фенилаланина в тирозин.
Коферментзависимая гиперфенилаланинемия (вариантная ФКУ) является следствием мутаций в генах, контролирующих метаболизм тетрагидробиоптерина.
Тетрагидробиоптерин необходим в реакциях гидроксилирования не только фенилаланина, но также тирозина и триптофана. Поэтому при дефиците этого кофермента нарушается в равной степени метаболизм всех 3 аминокислот. Заболевание характеризуется тяжелой неврологической симптоматикой и ранней смертностью («злокачественная ФКУ»).
Данное заболевание поддается фармакотерапии тетрагидробиоптерином.
![Схема точек приложения тирозиназы [http://www.cnb.uam.es/~montoliu/ albinomouse.html]. Тирозиназа необходима для синтеза меланина в меланоцитах и пигментном эпителии сетчатки.](/img/s/9/67/2206567_3.png)
Рис. 16.10. Схема точек приложения тирозиназы [http://www.cnb.uam.es/~montoliu/ albinomouse. html]. Тирозиназа необходима для синтеза меланина в меланоцитах и пигментном эпителии сетчатки.
Альбинизм — врождённый дефицит или отсутствие пигмента в коже, волосах, радужке и сетчатке глаза или только в радужке глаза за счёт нарушения обмена тирозина при синтезе меланинов (отсутствие или дефицит тирозиназы). Принято различать генерализованные (кожно-глазные), изолированные (глазные) и смешанные типы альбинизма.

![Рис. 16.11. Альбиносы на фотографиях IX века [http://members.optusnet.com.au/~msafier/albinism/]. Очаги альбинизма были выявлены в Северной Ирландии, в Южной Панаме. За цвет кожи и ночной образ жизни альбиносов называли «детьми Луны».](/img/s/9/67/2206567_5.jpg)
Рис. 16.11. Альбиносы на фотографиях IX века [http://members.optusnet.com.au/~msafier/albinism/]. Очаги альбинизма были выявлены в Северной Ирландии, в Южной Панаме. За цвет кожи и ночной образ жизни альбиносов называли «детьми Луны».
Нарушение обмена аминокислот. Патология конечных этапов белкового обмена, роль печени и почек в метаболизме аммиака. Нарушение обмена пуриновых и пиримидиновых оснований.