Растворы полимеров.
Методы исследования современных полимерных материалов

Принцип метода ГПХ. При фракционировании этим методом через колонку, заполненную частицами пористого сорбента в растворителе, пропускают раствор полидисперсного полимера, при этом молекулы стремятся диффундировать в растворитель, который находится в порах, т. е. проникают в поры. При постоянном потоке растворителя растворенное вещество движется вдоль колонки, и макромолекулы будут проникать… Читать ещё >
Растворы полимеров. Методы исследования современных полимерных материалов (реферат, курсовая, диплом, контрольная)
Молекулярно-массовое распределение является одной из важнейших характеристик высокомолекулярных соединений, которая отражает кинетический процесс полимеризации и определяет эксплуатационные характеристики полимеров, предсказывая пути его переработки. Рассмотрим в данном разделе понятие молекулярно-массового распределения полимеров и возможные методы их фракционирования.
Молекулярно-массовые характеристики полимеров
Основными молекулярно-массовыми характеристиками полидисперсных полимеров являются средние молекулярные массы (ММ), функции молекулярно-массового распределения (ММР) и кривые распределения, соответствующие этим функциям.
Для того, чтобы количественно охарактеризовать распределение полимера по ММ, необходимо рассчитать относительное количество фракций, содержащих макромолекулы одинаковой ММ. Это можно сделать двумя способами — исходя из числа или суммарной массы макромолекул. имеющих ММ, равную Mi; ni Mi — общая масса полимера.
Средняя ММ полидисперсного полимера является средневзвешенной величиной, вклад в которую каждой из фракций определяется ее ММ и относительным количеством. Из последнего следует, что для полидисперсного полимера характерны две средние ММ — среднечисловая ММ M n :
Из выражения (3) следует, что среднечисловая ММ равна общей массе макромолекул, деленной на их число.
При экспериментальном изучении ММР обычно имеют дело с непрерывными кривыми и функциями распределения. Значение непрерывной дифференциальной числовой функции распределения fn(M) равно числовой доле макромолекул с ММ от M до M+dM, деленной на dM; значение непрерывной массовой функции распределения fw(M) равно массовой доле макромолекул с ММ от M до M+dM, деленной на dM.
Непрерывные дифференциальные числовые и массовые функции связаны между собой, как и соответствующие дискретные функции, простым соотношением:
fw (M) (M M n) fn (M)(5).
Помимо дифференциальных, широко используются интегральные функции распределения:
значение (ордината) интегральной числовой функции распределения Fn(М) равно числовой доле макромолекул, имеющих ММ от минимальной до заданной М;
значение (ордината) интегральной массовой функции распределения Fw(М) равно массовой доле макромолекул, имеющих ММ от минимальной до заданной М.
Важной характеристикой полидисперсного полимера является ширина ММР. Отношение M w M n, которое характеризует ширину ММР, называют коэффициентом полидисперсности Шульца. Часто в соотношениях, характеризующих ММР полимера, вместо ММ используется степень полимеризации p M M 0, где М и М0 — молекулярные массы полимера и мономера.
Средние молекулярные массы M n, и M w определяются с помощью абсолютных методов, так как их вычисления проводятся без всяких предположений о форме и размерах макромолекул. Среднечисловая молекулярная масса M n может быть определена любым методом, основанным на измерении коллигативных (т.е. зависящих только от числа частиц) свойств растворов полимеров: осмометрия, эбуллиоскопия, криоскопия, изотермическая перегонка, измерение тепловых эффектов конденсации, а также по данным количественного определения концевых функциональных групп макромолекул какими-либо физическими или химическими методами. Среднемассовое значение M w, можно определить, например методом светорассеяния.
Практически кривые ММР получают в результате фракционирования полимеров, т. е. осуществляя различными методами разделения образца полимера на фракции с разными молекулярными массами.
Методы фракционирование полимеров
Разделение полимера на фракции основано на том, что критические температуры растворения полимеров зависят от их молекулярной массы. Не смотря на то, что раствор полимера представляет собой многокомпонентную систему (из-за наличия в полимере макромолекул с разными ММ) можно его рассматривать как квазибинарную систему, поскольку при фазовом расслоении такой многокомпонентной системы обычно наблюдается образование и сосуществование только двух фаз.
Составы фаз, образующихся при расслоении раствора полимера, неодинаковы и могут быть определены из условия равновесия z z, означающего, что химические потенциалы z-мера в сосуществующих фазах одинаковы. Пусть z относится к более концентрированной фазе, а — к более разбавленной фазе. Выражения для и z могут быть получены из уравнения для изобарно-изотермического потенциала раствора полимераНеобходимо учесть, что полимер полимолекулярный, и принять параметр независящим от ММ полимера.
- — при любом значении для разных z-меров соотношение z z будет различно, т. е. разделение фаз сопровождается факционированием полимера;
- — обычно > 0 и я > z, т. е. любые z-меры независимо от z присутствуют в
более концентрированной фазе, т. е. макромолекулы низкой ММ всегда содержатся в высокомолекулярной фракции каждая фракция имеет свое ММР, конечно, более узкое, чем исходный полимер;
добавлением осадителя к раствору полимера;
- — испарением растворителя, если полимер был предварительно растворен в смеси растворитель-осадитель;
- — изменением температуры раствора, которое приводит к ухудшению качества растворителя.
Метод фракционного растворения состоит в последовательном экстрагировании полимера серией жидкостей, растворяющая способность которых по отношению к данному полимеру последовательно возрастает. Исходный полимер может быть твердым, в виде коацервата, пленки, на инертном или активном носителе. Получаемы фракции обладают последовательно возрастающей молекулярной массой.
К аналитическим методам фракционирования относятся: ультрацентрифугирование, турбидиметрическое титрование, гель-проникающая хроматография и т. д.
Турбидиметрическое титрование заключается в измерении мутности раствора полимера при добавлении к нему осадителя. Если раствор полимера достаточно разбавлен, то макромолекулы полимера, выделяющегося при добавлении осадителя, остаются в виде устойчивой суспензии, обуславливающей мутность раствора. По мере добавления осадителя мутность раствора растет до тех пор, пока не выделится весь полимер, после чего мутность остается постоянной. Результаты титрования представляют в виде зависимости оптической плотности раствора, которая пропорциональна мутности, от объемной доли осадителя. В этом методе есть два главных допущения:
— принимается, что количество осадителя, необходимое для начала осаждения полимера (критический объем осадителя или порог осаждения) зависит от концентрации полимера в момент осаждения (С) и его молекулярной массы (М) по уравнению:
кр k lg C f M (10),.
где кр — объемная доля осадителя при пороге осаждения, k — константа, f M ;
некоторая функция ММ значение которой определяют из калибровочных титрований узких фракций полимера с известными ММ;
— полагают, что мутность пропорциональна количеству осажденного полимера и при добавлении небольшого количества осадителя () увеличение мутности () связано только с выделением макромолекул определенной длины z.
Ценность метода — быстрота и возможность работать с очень малыми количествами полимера (несколько мг). Метод оказывается очень полезным при подборе систем осадитель-растворитель для препаративного фракционирования, при определении пределов растворимости сополимеров, для качественной оценки ММР полимеров при изучении механизма полимеризации и т. д.
Фракционирование по методу гель-проникающей хроматографии
осуществляется по принципу молекулярного сита (хроматографическое разделение молекул происходит только по размеру, за счет их различной способности проникать в поры, и не зависит от химической природы компонентов). Этот метод объединяет в себе непрерывное фракционирование образца, которое основывается на различии в межфазном распределении веществ, движущихся вместе с растворителем (подвижная фаза) сквозь высокодисперсную среду неподвижной фазы и последующий анализ фракций. Принципиальной особенностью метода является возможность разделения молекул по их размерам в растворе в диапазоне молекулярных масс от 102 до.
108, что делает его незаменимым для исследования синтетических и биополимеров.
Принцип метода ГПХ. При фракционировании этим методом через колонку, заполненную частицами пористого сорбента в растворителе, пропускают раствор полидисперсного полимера, при этом молекулы стремятся диффундировать в растворитель, который находится в порах, т. е. проникают в поры. При постоянном потоке растворителя растворенное вещество движется вдоль колонки, и макромолекулы будут проникать в поры лишь тогда, когда их концентрация вне пор окажется больше чем в порах. Когда зона растворенного вещества покидает данный участок сорбента, концентрация образца внутри сорбента становится больше, чем вне его, и макромолекулы вновь диффундируют в поток подвижной фазы. Этот процесс повторяется циклически по всей длине колонки. В полидисперсном полимере всегда есть доля «коротких» макромолекул, которые легко проникают во все поры сорбента, доля более «крупных» макромолекул, способных проникать лишь в часть пор и «большие» макромолекулы, которые не проникают в поры совсем, являются «запретными» для пор данного размера. В соответствии с разной способностью к проникновению в поры, макромолекулы удерживаются в колонке разное время: первыми из колонки вымываются «запретные» большие макромолекулы, последними — самые маленькие. Такое разделение в хроматографической колонке называется методом «пространственного запрета». Время, в течение которого полимерные молекулы удерживаются в порах, называется временем удерживания tr. Т. е. это среднее время прохождения макромолекулы по колонке с момента ввода пробы до некоторого расстояния, равного длине колонки.
Время удерживания tr является основной экспериментально определяемой характеристикой хроматографического процесса.
Еще один параметр, которым наиболее часто пользуются в хроматографии.
— «удерживаемый объем» — Vr, который связан с временем удерживания:
Vr = tr v (11),.
где v — объемная скорость протекания растворителя через колонку (мл/мин), которая задается в начале эксперимента.
Удерживаемый объем — это количество миллилитров растворителя, которое необходимо пропустить через колонку для вымывания образца из пор сорбента. Эта величина связана с размером макромолекул аналогично времени удерживания: Vr минимален для «запретных» молекул и максимален для полностью проникающих в поры макромолекул.
Объем эксклюзионной колонки можно выразить суммой трех слагаемых:
V = Vо + Vs + Vd (12),.
где Vо — «мертвый» промежуточный объем колонки, т. е. объем растворителя, между частицами сорбента (объем подвижной фазы); Vs — объем пор, занятый растворителем (объем неподвижной фазы); Vd — объем матрицы сорбента без учета пор.
Полный объем растворителя в колонке Vt (полный объем колонки), представляет собой сумму объемов подвижной и неподвижной фаз.
Равновесный коэффициент распределения макромолекул между подвижной и неподвижной фазами k характеризует вероятность диффузии макромолекул в поры и зависит от соотношения размеров молекул и пор, а также определяет удерживание молекул в эксклюзионной колонке:
k = Сs/Cо (14),.
где Сs — концентрация вещества в неподвижной фазе; Cо — в подвижной фазе. Так как подвижная и неподвижная фазы имеют одинаковый состав,.
то k вещества, для которого обе фазы одинаково доступны, равен 1. Эта ситуация реализуется для молекул с самыми малыми размерами (в том числе и молекул растворителя), которые проникают во все поры и поэтому движутся через колонку наиболее медленно. Их удерживаемый объем равен полному объему растворителя.
Все молекулы, размер которых больше размера пор сорбента, не могут попасть в них (полная эксклюзия) и проходят по каналам между частицами. Они элюируются из колонки первыми с одним и тем же удерживаемым объемом, равным объему подвижной фазы (Vо). Коэффициент распределения k для этих молекул равен 0.
Молекулы промежуточного размера, способные проникать только в какую-ту часть пор удерживаются в колонке в соответствии с их размером. Коэффициент распределения k этих молекул изменяется от 0 до 1 и характеризует долю объема пор, доступных для молекул данного размера. Их удерживаемый объем определяется суммой Vо и доступной части объема пор:
Этоосновное.
Vr = Vо + k Vs (15).
Удерживание в уравнение, описывающее хроматографическом процессе.
Удобство метода ГПХ состоит в том, что основной параметр метода — удерживаемый объем Vr является однозначной функцией молекулярной массы (М).
В общем случае зависимость Vr (М) выражается:
lg M = C1 — C2Vr +С3V2r + (16).
Определение распределения по объему элюента с (Vr) и калибровочной кривой Vr (М) позволяет легко получить интегральное и дифференциальное молекулярно-массовое распределение:
Vr (М).
M
Fw (M) F(Vr)с(Vr)dVr
fwdM1 (17),.
Vr (M1).
M1.
где М1 — молекулярная масса самого низкомолекулярного компонента.
Т.е переход от интегрального распределения по объему элюента F (Vr) к интегральному распределению F (M) сводится к замене оси абсцисс (Vr) на ось lgM по уравнению lg M = C1 — C2Vr.
Т.о. калибровочная процедура заключается в последовательном хроматографировании стандартов и определении удерживаемых объемов Vr максимумов пиков. Полученные для каждого стандарта Vr наносят на график lgМпика = f (Vr) и соединяют плавной линией, которая и является калибровочной кривой, которую далее используют при анализе полимеров с неизвестной молекулярной массой.
Разбавленные растворы
Поведение макромолекул в растворе существенным образом зависит от термодинамического качества растворителя, молекулярной массы полимера и температуры раствора. Изменение этих параметров сказывается на размерах и форме макромолекулярных клубков, что приводит к изменению гидродинамических свойств разбавленных растворов полимеров. Основным источником информации о молекулярных характеристиках полимеров является изучение их свойств в разбавленных растворах методами молекулярной оптики и гидродинамики. Прежде всего, это методы статического и динамического светорассеяния, применение которых позволяет определить ММ, размеры и конформацию растворенных объектов. Следует отметить, что использование данных методов особенно продуктивно при исследовании свойств полимерных надмолекулярных структур, поскольку дает информацию о системе, неподверженной внешним воздействиям. Другой абсолютный метод ;
седиментационно-диффузионный анализ позволяет надежно измерять константы седиментации S и коэффициенты поступательной диффузии D в области сильного разбавления полимерных растворов. Полученные величины дают возможность определить ММ и гидродинамический радиус Rh исследуемых полимеров.
Метод светорассеяния
Принцип метода светорассеяния. Метод светорассеяния основан на эффекте рассеяния части света, проходящего через жидкую среду. Это рассеяние обусловлено наличием флуктуаций плотности и концентрации вещества в отдельном объеме раствора, существующих вследствие теплового движения. Различие в плотностях приводит к появлению различий в показателях преломления. Свет, проходящий через жидкую среду, преломляется на границах участков с различной плотностью, отклоняется от первоначального направления, т. е. рассеивается. Рассеяние тем больше, чем больше флуктуация. Если среда представляет собой раствор полимера, то, кроме флуктуаций плотности растворителя, имеют место флуктуации концентрации полимера. Эти флуктуации тем интенсивнее, чем меньше осмотическое давление внутри участков с большей концентрацией, т. е. чем больше ММ растворенного вещества.
Основное применение получили методы, основанные на измерении интенсивности света, рассеянного растворами полимеров, и ее угловой зависимости. Различают методы статического рассеяния света (упругого) или динамического (квазиупругого) рассеяния. Основное различие состоит в методе измерения интенсивности рассеянного света.
При упругом, или Рэлеевском рассеянии падающий и рассеянный свет имеют одну и ту же длину волны. В результате движения центров рассеяния рассеянный свет перестает быть монохроматическим, и вместо одной линии наблюдается рэлеевский пик. В этом случае измеряется усредненная во времени общая интенсивность как функция угла рассеяния. Используя статическое светорассеяние, можно определить массу и характерный линейный размер частиц в некоторых системах.
При неупругом рассеянии частота рассеянного света отличается от частоты падающего света. В методе динамического рассеяния света измеряется изменение интенсивности рассеяния во времени. Откуда определяют коэффициенты диффузии частиц, размер частиц и распределение по размерам.
Рэлей при изучении простейшего случая рассеяния в идеальном газе низкой плотности в 1871 г. предложил теорию статического (упругого) рассеяния. Он рассматривал рассеяние света сферическими диэлектрическими частицами с диаметром d, много меньшим длины волны л падающего света (d <? 20), и с коэффициентом преломления, близким к единице. В отсутствие поглощения и при использовании неполяризованного света уравнение Рэлея имеет следующий вид:
Молекулярная теория рассеяния света жидкостями разработана Эйнштейном. Рассматривая рассеяние как следствие термических флуктуаций плотности, он получил уравнение для мутности жидкости:
Дебай применил теорию Эйнштейна к растворам полимеров. В разбавленных растворах полимеров (с < 0.5 г/дл) рассеивающими центрами являются полимерные клубки. Если молекулярные клубки малы (h 2 1 2 400 A) по сравнению с длиной волны падающего света (т.е. составляет 0.05 + 0.1л), то интенсивность рассеянного света не зависит от угла рассеяния света и угловая зависимость — шар (рис.1). В этом случае справедливо уравнение Дебая.
— инкремент показателя преломления, Rи — число Рэлея или коэффициент рассеяния связанный с мутностью раствора.
Подставив в уравнение Дебая выражение для осмотического давления величину второго вириального коэффициента (Рис. 1, а).
а б Рис. 1. Интенсивность рассеянного света не зависит от угла рассеяния (а);
концентрационные зависимости обратной интенсивности рассеянного света Кс/R =90 для полистиролов, полученных реакцией передачи цепи с трис-(пентафторфенил)германом с различной молекулярной массой [(C6F5)3GeH]=0.02 моль/л (1) и [(C6F5)3GeH]=0.005 моль/л (2) в хлороформе от концентрации (б) Для определения ММ полимеров, размер макромолекул которых мал по сравнению с длиной волны (меньше л/20), достаточно найти величину относительного избыточного рассеяния под углом 90. Найденная методом светорассеяния ММ соответствует средневесовому значению. Все приведенное выше относится к малым частицам (по сравнению с длиной волны падающего света). С увеличением размеров изменяются закономерности светорассеяния, появляется сферическая ассиметрия интенсивности рассеянного света, изменяется степень его поляризованности. Различия при рассеянии на малых и крупных частицах (? л/20) могут быть продемонстрированы с помощью векторных диаграмм Ми (Рис. 2).
Рис. 2. Диаграмма Ми для, а 0 малых (а) и больших (б) частиц. Заштрихованная часть соответствует степени поляризации рассеянного света.
Для больших макромолекул интенсивность рассеянного света сферически нессиметрична. Это происходит вследствие того, что электромагнитные колебания, которые возбуждаются на удаленных друг от друга участках макромолекулы, оказываются не в фазе. Разница фаз волн двух индуцированных диполей оказывается тем больше, чем больше размеры макромолекул и чем больше угол рассеяния и (Рис. 3).
В направлении первичного светового пучка разность фаз равна нулю, а в обратном направлении — наибольшая. Угловая зависимость интенсивности рассеянного света — индикатрисса рассеяния — используется для определения размеров и форм макромолекул в растворах. В связи с этим в уравнение Дебая вводится фактор внутренней интерференции (или форм-фактор — Pи): свeт, который рассeиваeтся.
Рис. 3. Рассеяние света макромолекулой Для макромолекул любой формы Pи = 1 при и = 0 С. С увеличением и величина Pи уменьшается. Форм-фактор связан с радиусом инерции и зависит от формы частиц (сферы, цилиндры, тонкие палочки и т. д.) В соответствии с уравнением Зимма форм-фактор для частицы с конфигурацией статистического клубка равен:
1Р 1 (8 2 9 2)h 2 sin 2 (/ 2) (26),.
где h 2 средний квадрат расстояния между концами полимерной цепи.
Подставляя уравнение (26) в (25) получаем:
Средний квадрат расстояния между концами полимерной цепи, определяемый из уравнения (28) является z — средней величиной. Зная h 2 ,.
можно рассчитать среднеквадратичный радиус инерции r 2 1 2 (для линейных полимеров), используя следующее соотношение:
h 2 6r 2 (29).
Для определения ММ и других параметров больших молекул, когда наблюдается угловая ассиметрия светорассеяния, обычно используютметодику двойной экстраполяции (или методику Зимма).
Зимм показал, что зависимости:
- — KcR от с, при и = const;
- — KcR от sin2(и/2), при с = const;
- — KcR c 0 от sin2(и/2) и KcR c 0 от с представляют собой прямые линии.
Т.е. прямые, описываемые уравнениями (30) и (31), отсекают на оси ординат отрезок, равный 1/Mw. По величине соответствующих угловых коэффициентов могут быть найдены значения A2 и h 2. Описанная экстраполяция может графически изображена на одной диаграмме называемой диаграммой Зимма (Рис. 4) представляющей собой зависимость KcR от (100с2+sin (и/2). Для получения этой диаграммы измеряют рассеяние света раствором одной концентрации под разными углами и, строят график зависимости в приведенных на рис. 4 координатах, экстраполируя приведенную зависимость к и= 0. Затем получают серию таких зависимостей для разных концентраций. Через точки, отвечающие одному углу и разным концентрациям, проводят линии, которые затем экстраполируют к с=0. Линии соответствующие и = 0 и с = 0, пересекаются в одной точке (Б), отсекая на оси ординат отрезок, по которому находят средневесовую ММ полимера. Из наклона линии БС0 при с=0 можно определить радиус инерции макромолекулы, а из наклона линии Би0 при и=0 — второй вириальный коэффициент.
Рис. 4. Общий вид диаграммы Зимма.
Необходимым условием применения методов светорассеяния для изучения растворов полимеров является отсутствие многократного рассеяния, т. е. каждый фотон падающего излучения должен испытать взаимодействие только с одной молекулой полимера в растворе. Это достигается соответствующим разбавлением системы и уменьшением объема исследуемого образца.
В последнее время для определения молекулярно-массовых характеристик широкое развитие получили так называемые «транспортные» методы, основанные на изучении макроскопического переноса вещества в жидкой среде под действием внешней силы. В основе этих методов лежит зависимость транспортной подвижности макромолекул от их молекулярной массы. К таким методам относятся седиментация, диффузия, электрофорез и хроматографическое разделение макромолекул.
Седиментация и диффузия
Диффузия непосредственно связана с подвижностью молекул, следовательно, ее скорость должна зависеть от их размеров. Количественная связь между коэффициентом диффузии D и размером диффундирующей частицы была получена Эйнштейном:
Это уравнение справедливо и для диффундирующих коллоидных частиц, и для молекул. Если молекула имеет шарообразную форму, то ее объем V 4 r3 .
Умножив эту величину на плотность d, получаем массу молекулы, а при умножении на число Авогадро NA, получаем массу 1 моля, т. е.
Выразив r из уравнения Эйнштейна (32), и подставив его в уравнение (33), получим выражение для молекулярной массы шарообразной молекулы.
отношение коэффициентов диффузии шарообразной и нешарообразной частиц, движущихся с одинаковой скоростью.
Все имеющиеся уравнения несправедливы для диффузии макромолекул, и в настоящее время не существует уравнения, связывающего коэффициент поступательной диффузии макромолекул с их ММ. Поэтому ММ рассчитывают из эмпирических соотношений. Для этого определяют коэффициент поступательной диффузии макромолекул, используя второй закон Фика:
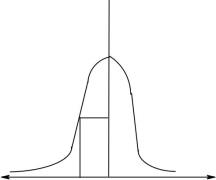
Методы определения коэффициента диффузии основаны на измерении скорости размывания границы между раствором исследуемого полимера и растворителем. Распределение концентрации и градиента dcdx около границы при различных временах наблюдения выражается кривыми, приведенными на рисунке 5. Распределение dcdx имеет вид гауссовой кривой с максимумом на границе между раствором и растворителем (x=0). Оно описывается уравнением Винера, полученным интегрированием второго закона Фика при определенных допущениях:
где с0 — концентрация раствора, г/см3; t — время от начала диффузии, с; x — расстояние рассматриваемого градиента от границы.
в.
Рис. 5. Кривые распределения концентрации (а), градиента концентрации (б) и градиента показателя преломления (в) Для регистрации обычно пользуются оптическими методами, например, определяют коэффициент преломления n раствора и его градиент dndx вдоль направления диффузии.
Для рабочих концентраций (0.5 г/мл), используемых в этом методе, градиент показателя преломления прямо пропорционален градиенту концентрации:
После интегрирования уравнения (37) в пределах от n0 до n1 (где n0 и n1 — показатели преломления растворителя и раствора) получаем:
Распределение градиента коэффициента преломления также выражается гауссовой кривой (рис. 5, в).
Наиболее простой метод расчета — метод максимальной ординаты, которая отвечает x = 0. При этом выражение x2 4Dt в уравнении (39) обращается в нуль, и это уравнение упрощается:
Метод седиментации. Как известно, зная скорость оседания частиц можно определить их размеры. Оседание молекул в центробежном поле происходит в направлении, перпендикулярном оси вращения. При оседании молекулы (объемом v) под действием центробежной силы постоянно изменяется ее расстояние от оси вращения (x). При этом центробежная сила зависит от угловой скорости вращения центрифуги (w2x). Сила сопротивления выражается законом Стокса (для сферических частиц):
Коэффициент диффузии определяется, как было показано в методе диффузии. Константа седиментации определяется при помощи ультрацентрифуги. Для этого через кювету с раствором полимера, помещенную в ультрацентрифугу пропускают пучок света, который падает на фотопластинку, находящуюся за кюветой. При вращении кюветы по мере осаждения вещества граница раздела между раствором и растворителем постепенно перемещается, и свет поглощается по высоте кюветы в различной степени. На фотопластинке получаются полосы разной степени почернения. Фотометрируя снимки, сделанные через определенные промежутки времени, можно получить седиментационную кривую, т. е. кривую распределения градиента концентрации вдоль высоты кюветы при разных временах.
Зависимость lnx от t должна выражаться прямой, по углу наклона которой можно рассчитать коэффициент седиментации S.
Для исключения концентрационных эффектов находят значение S0, экстраполируя величину S на бесконечное разбавление (т.е. строя график зависимости 1S f (c)). Зная значения S0 и D0 находят молекулярную массу полимера по формуле (44).
Метод седиментации в ультрацентрифуге является абсолютным методом измерения молекулярной массы полимера, т.к. в нем не делается никаких предположений о конформациях макромолекул.
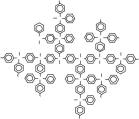
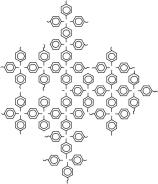
В сочетании методы светорассеяния, вискозиметрии и седиментационно-диффузионного анализа могут дать полную информацию о гидродинамических и конформационных свойствах макромолекул в растворе, что особенно важно для сверхразветвленных полимеров. Методами молекулярной гидродинамики и оптики в разбавленных растворах в хлороформе нами были исследованы две серии сверхразветвленных сополимеров различной топологической структуры на основе перфторированных гидридов германия (ФГ и ДГ). Варьируя количество ДГ, и его последовательность введения в мономерную смесь в ходе синтеза удалось получить полимеры с различной архитектурой. Первая серия — сополимеры различной молекулярной массы (от 2.3· 104 до 31· 104) с жесткими линейными цепями между точками ветвления и разным числом точек разветвления в каскадах дендритного фрагмента, вторая — сополимеры, которые при близкой степени ветвления в среднем имеют более «рыхлую» структуру за счет большего числа линейных звеньев на переферии макромолекул с молекулярной массой от 2.5· 104 до 23· 104 (Рис. 6).
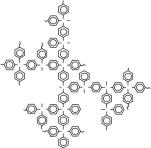
в).
Рис. 6. Схематическая структура (со)полимеров на основе трис-(пентафторфенил)герман и бис-(пентафторфенил)германа различной архитектуры: сополимеры с жесткими линейными цепями между точками ветвления и разным числом точек разветвления в каскадах дендритного фрагмента (а), сополимеры, которые при близкой степени ветвления в среднем имеют более «рыхлую» структуру за счет большего числа линейных звеньев на переферии макромолекул (б) и сверхразветвленный перфторированный полифениленгерман (в).
Исследованные сополимеры имеют очень низкие характеристические вязкости. Значения [ ] изменяются от 1.5 до 5 см3/г при увеличении ММ от 1.4· 104 до 25· 104. Для сверхразветвленных полимеров с гибкими цепями между точками ветвления при таких ММ обычно получают [ ] в интервале от 3 до 50 см3/г. Низкие значения [ ] для со-ПФГ однозначно указывают на компактные размеры их макромолекул, на высокую плотность полимерного вещества в объеме, который они занимают в растворе. В соответствии с соотношением Эйнштейна для твердой сферической частицы [ ]сф = 2.5 v. Подставляя, значение парциального удельного объема для со-ПФГ, имеем [ ]сф 1.3 см3/г, что несколько меньше величины характеристической вязкости для самого низкомолекулярного полимера — ПФГ (таблица 1), полученного в отсутствии бис-(пентафторфенил)германа. Соответственно можно предположить, что форма его макромолекул очень незначительно отличается от сферической. Из таблицы 1 видно, что для ПФГ отношение [ ]/[ ]сф 1.2. Такое низкое значение [ ]/[ ]сф характерно скорее для дендримеров, чем для сверхразветвленных полимеров. Для последних обычно [ ]/[ ]сф 1.5.
Таблица. | 1. | Молекулярно-массовые. | и. | гидродинамические. | характеристики. | |||||||||||
сверхразветвленных сополимеров на основе ФГ и ДГ. | ||||||||||||||||
№. | xДГ,. | [ ],. | Mw. | R | ДРС. | . | S0,. | MSD · 10. | — 4. | A0· 1010,. | ||||||
Полимер | · 10. | — 4. | R, нм. | |||||||||||||
п/п. | вес.%. | см3/г. | h D | Св. | Да. | |||||||||||
Да. | нм. | эрг/град/моль1/3. | ||||||||||||||
ПФГ. | 1.5. | 1.3. | 2.2. | 2.0. | 1.6. | 1.5. | 2.0. | |||||||||
со-ПФГ-а. | 2.4. | 2.3. | 3.5. | 3.2. | 2.5. | 2.1. | 1.8. | |||||||||
2.3. | 5.0. | 4.4. | 3.3. | 4.2. | 2.6. | 1.6. | ||||||||||
2.6. | 5.9. | 5.7. | 4.4. | 6.4. | 2.9. | 2.1. | ||||||||||
2.9. | 7.7. | 5.9. | 5.4. | 9.1. | 3.3. | 1.7. | ||||||||||
3.7. | 9.9. | 9.8. | 5.6. | 1.6. | ||||||||||||
со-ПФГ-б. | ; | 2.5. | 3.4. | 1.5. | 1.6. | ; | ; | |||||||||
2.5. | 3.2. | 3.6. | 3.2. | 2.6. | 2.3. | 0.8. | ||||||||||
2.6. | 4.3. | 3.7. | 4.3. | 3.9. | 2.6. | 2.1. | ||||||||||
2.8. | 5.8. | 7.1. | 3.4. | 5.5. | 2.9. | 1.2. | ||||||||||
4.6. | 9.0. | 9.2. | 5.3. | 1.7. | ||||||||||||
По полученным данным можно сделать вывод о том, что макромолекулы первой серии (Рис. 6, а) характеризуются несколько более компактными размерами и меньшей асимметрией формы по сравнению с молекулами второй серии (Рис. 6, б). Вероятно, это связано с тем, что сополимеры первой серии имеют в своем составе димерную цепь из звеньев ДГ на которую «нанизаны» разветвленные блоки, такие молекулы более жесткие и по форме напоминают слабовытянутый эллипсоид, в отличие от сверхразветвленных макромолекул второй серии, для которых характерно наличие жесткого сферического ядра с фокальной точкойGe (C6F5)3 и ветвями из звеньев ДГ и ФГ, что обеспечивает в целом более рыхлую структуру по сравнению со сверхразветвленным ПФГ Т.о. методами молекулярной гидродинамики и оптики показано, что макромолекулы исследованных полимеров имеют компактные размеры и характеризуются высокой плотностью полимерного вещества, а асимметрия их формы невысока. По этим характеристикам они приближаются к дендримерам. При фиксированной молекулярной массе сополимеры с «рыхлой» структурой характеризуются большими размерами макромолекул и более высокими значениями характеристической вязкости.
Концентрированные растворы полимеров
Концентрированными называются растворы, в которых молекулы растворенного вещества взаимодействуют друг с другом. Интерес к реологическим свойствам таких растворов обусловлен, прежде всего, технологией переработки полимеров, многие из которых перерабатываются через растворы и расплавы.
Рассмотрим особенности механических свойств полимеров, находящихся в текучем состоянии. Под полимерами, находящимися в текучем (или вязкотекучем) состоянии обычно имеют в виду концентрированные растворы полимеров, расплавы кристаллизующихся полимеров и аморфные полимеры в таких режимах деформации и при таких температурах, когда определяющую роль в полной их деформации играет деформация вязкого течения, т. е. необратимая составляющая полной деформации.
Кроме основных методов термомеханического исследования, для текучих полимеров применим специфический метод, заключающийся в измерении напряжений в режиме постоянной скорости деформации. Хотя, в принципе, осуществить постоянную скорость деформации можно и при механических испытаниях твердых и высокоэластических полимеров, только для текучих полимерных систем этот метод приобретает решающее значение, т.к. только для них полная деформация может быть неограниченно велика и поэтому наблюдение за развитием напряжения в режиме постоянной скорости деформации может завершиться достижением режима установившегося течения. Этому режиму течения отвечают свои характерные значения напряжения и накопленной и сохраняющейся в материале высокоэластической деформации; дальнейшее же развитие деформации происходит только через вязкое течение, когда состояние материала не меняется во времени.
Вязкость полимеров зависит от ММ, температуры, давления, а также от режима деформации (скорости деформации и напряжения). Для подавляющего большинства полимерных систем для зависимости скорости от напряжения при сдвиговом течении характерен эффект аномалии вязкости, заключающийся в уменьшении эффективной вязкости по мере увеличения напряжения сдвига. При растяжении, наоборот, по мере увеличения скорости деформации и напряжения продольная вязкость увеличивается. Вязкость полимеров в значительной степени зависит от температуры. Для области высоких температур, далеких от температуры стеклования полимера, кривая вязкости от температуры описывается экспоненциальной зависимостью, которая характеризует величину свободной энергии активации вязкого течения U. По мере возрастания ММ энергия активации становится независящей от ММ, т. е. по мере удлинения молекул их движение при течении имеет сегментальный характер (для осуществления единичного акта течения требуется перемещение только части молекулы). Энергия активации линейных полимеров зависит от строения элементарного звена, увеличиваясь по мере повышения жесткости цепи. Несмотря на то, что механизм и осуществление элементарного акта течения не зависят от длины макромолекулы в целом, абсолютные значения вязкости существенно зависят от ММ, поскольку для необратимого перемещения макромолекул необходимо, чтобы путем независимых перемещений отдельных сегментов произошло смешение центра тяжести макромолекулы. Чем выше ММ, тем больше число согласованных перемещений должно произойти для того, чтобы сместить центр тяжести макромолекулы. Отсюда следует, что зависимость вязкости от ММ складывается из двух участков. Первый — область низких значений ММ, где вязкость пропорциональна ММ и второй участок, это та область, где ММ оказывает существенное влияние на вязкость и начинает выполняться условие M 3.5 .
Появление высокоэластических деформаций возможно в полимерах не только в высокоэластическом, но и вязкотекучем состоянии. Их развитие в обоих случаях обусловлено одним и тем же механизмом — отклонением Для высокомолекулярных полимеров модуль высокоэластичности значительно возрастает по мере расширения ММР.
Так же, как и в полимерах, находящихся в высокоэластическом состоянии, в текучих полимерах, особенно содержащих твердый наполнитель, возможны эффекты обратимого разрушения их структуры, что приводит к тиксотропному изменению свойств системы. Для текучих полимеров из-за повышенной подвижности макромолекул, процессы тиксотропного восстановления протекают быстрее, чем для полимеров находящихся в высокоэластическом состоянии.
Изучение вязкоупругих свойств концентрированных растворов необходимо для получения ценной информации об их структуре, которая представляет собой пространственную флуктуационную сетку, образованную плотно упакованными агрегатами, или ассоциатами макромолекул, внутри которых находятся молекулы растворителя.
Если рассматривать полимеры сложной архитектуры, например, сверхразветвленные полимеры и дендримеры, то с точки зрения общих проблем реологии, сферическая структура макромолекул позволяет рассматривать их, с одной стороны, как полимеры, а с другой — как коллоидные дисперсии.
Подробных исследований реологических свойств полимеров разветвленного строения в блоке к настоящему времени практически не проводилось, хотя именно детальное изучение вязкоупругих свойств этих полимеров, свободных от растворителей, может дать полезную информацию о специфике их поведения при течении как ансамбля коллоидных частиц с гибкими макромолекулярными фрагментами. К настоящему времени основные исследования реологических (в основном вязкостных) свойств были выполнены для разбавленных растворов дендримеров с целью выявления их молекулярного строения. В одной из работ (Hawker C.J. с сотрудниками), посвященных изучению реологических свойств дендримеров в расплаве, на примере дендритного полиэфира была обнаружена необычная зависимость вязкости от ММ. Для низших генераций дендримеров показатель степенной зависимости вязкости от ММ значительно больше единицы, а затем при достижении ММ порядка 104 (начиная с пятой генерации) эта зависимость становится линейной. Кривые течения дендримеров разных генераций полибензилового эфира носят ньютоновский характер в широкой области скоростей сдвига (0.1−100 с-1), что нехарактерно для полимеров. Другая ситуация имеет место для генераций полиамидоаминных дендримеров: при температурах, незначительно отличающихся от температуры стеклования, наблюдается снижение динамической вязкости при увеличении частоты сдвига (работы Wang H.).
Характер течения дендримеров и сверхразветвленных полимеров зависит от природы концевых групп. Так, модификация полипропилениминных дендримеров аминои цианогруппами привела к увеличению вязкости при сохранении ньютоновского поведения расплава. Tande B.M. со соавторами также было показано, что вязкость исходных полипропилениминных дендримеров четвертой и пятой генераций постоянна в широком диапазоне скоростей сдвига. В то же время для указанных дендримеров, модифицированных метили бензилакрилатом, наблюдается аномалия вязкости. Эти результаты демонстрируют важное влияние концевых групп на реологические свойства расплавов дендримеров. Можно было бы полагать, что при температурах выше точки стеклования уже возможно течение дендримеров. Однако авторами Смирновой Н. Н. и Терещенко А. С. с сотрудниками установлено, что для карбосилановых дендримеров высоких генераций помимо стеклования наблюдается второй, высокотемпературный переход. Он был обнаружен методом адиабатической калориметрии высокого разрешения, что было связано с характером межмолекулярных контактов в дендримерах. С целью детального исследования природы высокотемпературного перехода и возможного влияния на этот переход концевых групп дендримеров в широком температурном диапазоне были исследованы вязкоупругие свойства производных карбосиланового дендримера, различающихся типом концевых групп (Миронова М.В. и др.). Было показано, что карбосилановые дендримеры высоких генераций способны образовывать надмолекулярную структуру в виде физической сетки межмолекулярных контактов. Разрушение сетки может быть инициировано сдвиговой деформацией и температурой. Высокотемпературный переход в карбосилановых дендримерах высоких генераций, вызванный разрушением физической сетки, имеет релаксационную природу. Он определяется специфическим межмолекулярным взаимодействием концевых групп дендримеров и зависит от их подвижности. Наличие коротких силоксановых заместителей приводит к появлению высокоэластических свойств выше области стеклования, в то время как введение менее гибких карбосилановых и бутильных заместителей способствует проявлению типичных «полимерных» свойств. Таким образом, формируя ту или иную разновидность поверхностного слоя молекулярной структуры исходных дендримеров, можно регулировать их вязкоупругие свойства в широких пределах.
4. Методы исследования физико-химических и механических свойств полимерных материалов
Химическая структура полимеров, т. е. его химический состав и способ соединения атомов в макромолекуле, не определяет однозначно поведение полимерного материала. Свойства полимеров зависят не только от химической, но и от их физической (надмолекулярной) структуры. Структурные процессы изучают с помощью методов, которые основаны на измерении зависимости какого-либо показателя физических свойств полимерного материала от его структуры. Сюда можно отнести: методы термического анализа (измерение теплоемкости, температур переходов, дифференциальный термический анализ), зондовые методы (термодинамических параметров взаимодействия органических веществ с полимерами: коэффициенты растворимости, энтальпии сорбции, парциальные мольные энтальпии смешения, параметр растворимости, определение величины свободного объема в полимерах и др.), механические (измерение прочностных, деформационных и релаксационных свойств), электрические (диэлектрическая проницаемость, диэлектрические потери) и дилатометрические методы. Рассмотрим некоторые из перечисленных методов.
4.1 Методы термического анализа полимеров
К методам термического анализа относятся методы, по которым можно оценивать свойства полимеров в ходе изменения температуры (охлаждение или нагревание). Самыми распространенными являются дифференциальная сканирующая калориметрия (ДСК), термогравиметрический анализ (ТГА), динамический механический анализ (ДМА).
Дифференциальная сканирующая калориметрия. Это метод основан на измерении разницы тепловых потоков, идущих от испытуемого образца и образца сравнения, которые образуются в результате изменения физических или химических свойств исследуемого материала. Получаемая информация позволяет определять характер протекающих процессов и характеризовать свойства полимерного материала. Различие тепловых потоков возникает вследствие таких тепловых эффектов как плавление, кристаллизация, химические реакции, полиморфные превращения, испарение и др. В результате можно определить удельную теплоемкость и изменения теплоемкости, например в процессе стеклования полимера.
Термогравиметрический анализ. Это метод, в основе которого лежит постоянное взвешивание образца в зависимости от температуры при постоянной скорости нагревания в зависимости от времени. Он позволяет, используя небольшие количества вещества, получать информацию о кинетике и механизме деструкции полимера, его термостойкости, твердофазных реакциях, а также определять влагу, содержание остаточных материалов в полимере (мономер, растворитель, наполнитель), изучать процессы сорбции и состав композиционных полимерных материалов. Если к ТГА-анализатору подключить ИК-Фурье или масс-спектрометр, то анализ выделившихся газов даст полную информацию о механизме сложных термохимических процессах, которые идут в полимере при повышении температуры.
Динамический механический анализ применяется для исследований зависимости механических и вязкоупругих свойств (сдвиг, растяжение, сжатие, трехточечный и консольный изгиб) полимерных материалов от температуры, времени и частоты при воздействии периодических нагрузок. Детально этот метод анализа будет рассмотрен в разделе 4.3.
В настоящее время модернизация методов термического анализа привела к появлению модульных систем с уникальными техническими характеристиками, объединяющие методы ДМА, ДСК и ТГА. Это позволяет одновременно определять различные характеристики полимерного материала в широком диапазоне частот и температур, что позволяет получать информацию не только о механических свойствах (определяющих область применения полимера), но и о происходящих в материале молекулярных перегруппировках и возникающих структурах. Именно это открывает новые возможности для оптимизации выбора полимерного материала и процесса переработки, контроля качества, анализа разрушения полимера, изучения реакций сшивания полимеров, гелеобразования и др.
4.2 Транспортные и диффузионные методы (зондовые методы)
Область практического применения полимеров (например, защитные покрытия, мембраны, уплотнители) определяется их проницаемостью.
Механизмы проникновения газов через твердые тела Газопроницаемость полимеров, так же как и другие свойства, определяются такими факторами, как гибкость цепи; межмолекулярное взаимодействие; фазовое и физическое состояние полимера; плотность упаковки макромолекул; степень сшивания. Решающее значение для диффузионной проницаемости, которая обусловлена преимущественно сорбцией и диффузией, имеет гибкость цепи полимера и межцепное взаимодействие.
Сорбция газов полимерами. Газы могут адсорбироваться на внешней и внутренней поверхностях полимеров или растворяться в микропорах, возникающих между их макромолекулами. Общее количество поглощенного или сорбированного газа можно измерить, например, с помощью весов Мак-Бэна (чувствительные спиральные весы), и рассчитать концентрацию с газа в полимере. Она тем больше, чем больше парциальное давление p газа в окружающей среде: с p, где коэффициент пропорциональности.
называется коэффициентом сорбции (объем газа, поглощенный единицей объема полимера при парциальном давлении, равном единице, и температуре опыта). Коэффициент сорбции выражается в см3/(см3 кгс/см2). При сорбции газов и паров они могут конденсироваться в полимере, т. е. менять свое фазовое состояние, превращаясь в жидкость. В некоторых случаях сорбированное вещество может в полимере образовывать агрегаты или ассоциаты.
Для эластичного непоритсого полимера, в котором происходит только процесс растворения газа (газ заполняет свободный объем, который имеет флуктуационный характер, вследствие чего, молекулы газа при сорбции могут обмениваться местами со звеньями полимера), этот коэффициент называется коэффициентом растворимости газа, который зависит от парциального давления газа и температуры.
Процесс сорбции неинертных паров полимерами рассматривают как процесс взаимного растворения компонентов, который происходит по разным механизмам в зависимости от температуры:
- 1. Т Тс. Сорбирующий полимер находится в высокоэластическом состоянии, для которого характерна гибкость цепи и возможны перестановки молекул сорбата и звеньев или сегментов цепи полимера уже при очень малых значениях давления. Вид изотерм сорбции будет зависеть от термодинамического сродства сорбата к полимеру и гибкости цепи полимера. Чем меньше сродство, тем меньше сорбция.
- 2. Т Тс. Из-за отсутствия сегментального движения обмен между молекулами пара и звеньями цепи полимера затруднен. В этом случае молекулы пара могут проникать только в имеющиеся в полимере микропустоты, которые у плотноупакованных полимеров малы. Количество сорбированного вещества в этом случае мало, что лежит за пределами чувствительности сорбционного метода.
Поэтому процессы сорбции наиболее заметны, когда система находится в высокоэластическом состоянии, т. е. возможен обмен между молекулами сорбата и звеньями цепи, в результате чего, сначала происходит набухание полимера, которое может затем перейти в его растворение в парах сорбата.
Обращенная газовая хроматография
Для изучения термодинамики сорбции газов и паров в полимерах и определения их физико-химических параметров используют метод обращенной газовой хроматографии (ОГХ). Для этого полимер наносят на поверхность пористого твердого носителя, а сорбат вводят в поток газа-носителя. Метод ОГХ является весьма информативным для изучения термодинамики сорбции в полимерах, позволяющим также оценивать величину свободного объема.
Теоретические основы метода ОГХ. В методе ОГХ измеряется время удерживания сорбируемого tr и несорбируемого ta компонентов. Величина ta, соответствующая фактически почти не сорбируемому компоненту (обычно воздуху), необходима, чтобы учесть «мертвый» объем хроматографа. Таким образом, можно найти чистый удерживаемый объем VN: длине хроматографической колонки, и температуре T.
При приготовлении колонки, используемой в методе ОГХ, крайне важно знать массу (гр) введенной полимерной фазы wL. С учетом величины wL может быть получен удельный удерживаемый объем Vg: т. е. основная экспериментальная величина, от которой зависят все изучаемые термодинамические параметры.
Величина p0 (Па) — входное давление колонки, а параметр jnm представляет собой поправку, равную отношению давления, усредненного по времени пребывания пробы в хроматографической колонке, к давлению на выходе из нее.
Еще один термодинамический параметр, который может быть найден с бесконечном разбавлении:
Приведенный коэффициент активности характеризует отклонение от идеальности в бинарной системе полимер-пар (отклонение от закона Рауля для давления паров над раствором).
По температурной зависимости коэффициента активности можно вычислить парциальную мольную энтальпию смешения, характеризующую взаимодействие сорбата с полимером:
поскольку энтальпию сорбции можно представить как сумму Hs Hc Hm, где Hc — энтальпия конденсации различных сорбатов.
Эксперимент проводят на газовом хроматографе с детектором по теплопроводности. Температуру колонок необходимо поддерживать с точностью 0.5°С, давление на входе в хроматограф — до 0.6 кПа, что позволяет вводить поправки в уравнения (2) и (4), выходное давление приравнивалось к атмосферному. В качестве газа-носителя применяют гелий; его скорость измеряют с помощью мыльно-пленочного расходомера. Пик воздуха используется для определения ta.
Газохроматографический эксперимент начинают после стабилизации температуры и давления газа в колонке. Температуру испарителя устанавливают на 30 °C выше температуры кипения сорбата. Количество сорбата в пробе должно быть достаточным для идентификации пика на хроматограмме (~0.01 мкл). Для каждого сорбата при каждой температуре проводится по 7−10 параллельных опытов.
При изучении сорбции газов и паров в полимерах методом ОГХ крайне важно контролировать, устанавливается ли объемная сорбция во всем слое полимерной фазы в ходе хроматографического эксперимента. С этой целью в специальных опытах изучают влияние скорости газа-носителя (она варьируется от 2.5 до 30 см3/мин) на наблюдаемое время и объем удерживания. Например, при использовании н-октана при 45 °C и н-нонана при 50 °C на газовом хроматографе ЛХМ-80, было установлено, что при скорости ниже 5 см3/мин отсутствует заметное влияние скорости газа-носителя, поэтому все измерения в работе при разной температуре и с различными сорбатами проводили именно при этой скорости. Очевидно, что при более высокой температуре в ходе хроматографического эксперимента реализуется более высокий коэффициент диффузии. Об этом также свидетельствует линейность зависимости удельного удерживаемого объема от обратной температуры. Найденные таким образом термодинамические параметры соответствуют условиям бесконечного разбавления при объемной сорбции в полимере.
Давление насыщенных паров сорбатов находят с помощью уравнения Антуана и (или) уравнения.
lnPVP = AlnT + B/T + C + DT2,.
параметры которых табулированы в специальных Базах данных.
Если диаграммы удерживания сорбатов, т. е. зависимости логарифма удельного удерживаемого объема от обратной температуры во всем интервале изученных температур (30−115°C) линейны, то это свидетельствует о постоянстве внутренней энергии сорбции в изученном температурном интервале. По мере увеличения размера сорбируемой молекулы (ММ сорбата) при конкретной температуре происходит увеличение удельного удерживаемого объема. Линейный характер зависимостей, может быть еще одним подтверждением того, что в условиях эксперимента полностью устанавливается объемная сорбция в пленке полимера, нанесенного на носитель. Таким образом, измеренные значения Vg могут быть использованы для расчетов термодинамических параметров сорбции. По величинам Vg по формуле (4) рассчитывают коэффициент растворимости различных сорбатов в полимере. Диффузионные ограничения, приводящие к отклонениям от равновесной сорбции в хроматографическом эксперименте, являются причиной S-образной формы диаграмм удерживания.
Парциальная мольная энтальпия смешения, которая вычисляется по уравнению (6), проходит через минимум в зависимости от молекулярного размера сорбата, при этом координаты минимума соответствуют среднему размеру элемента свободного объема в стеклообразном полимере. Т. е. в процессе сорбции (при малых содержаниях растворенного вещества) молекулы сорбата заполняют преимущественно вакансии, имеющиеся в стеклообразном полимере (элементы свободного объема). В результате теплота смешения получается резко отрицательной, поскольку при этом молекулы сорбата не совершают работу по раздвижению полимерных цепей. Поэтому метод ОГХ с успехом применяется в качестве зондового для оценки величины свободного объема в стеклообразных полимерах, причем его результаты хорошо согласуются с данными других зондовых методов, например с методом аннигиляции позитронов.
Спектроскопия времен аннигиляции позитронов
Метод аннигиляции позитронов используется для получения информации о размерах и распределении по размерам элементов свободного объема, их концентрации, а также о влиянии на свободный объем таких факторов, как температура, давление, механические деформации и фазовый состав полимера. Используя этот метод можно проследить за изменениями свободного объема в процессе физического старения полимеров, в результате сорбции и набухания, в ходе сшивки. Параметры спектров времен аннигиляции позитронов сильно зависят от строения полимеров, и практически не зависят от ММ.
Этот метод основан на измерении времен жизни позитронов в веществе. В полимере позитроны могут существовать как в свободном состоянии (e+), так и в связанном. Последнее возможно в виде водородоподобного атома позитрония, т. е. электрон-позитронной парой (Ps или e-e+). Синглетное состояние этой частицы p-Ps имеет антипараллельные спины и короткое время жизни (0.125 нс в вакууме), в то время как триплетное состояние (o-Ps) с параллельными спинами имеет существенно большее время жизни (142 нс в вакууме). Считается, что долгоживущая частица o-Ps попадает в область с пониженной электронной плотностью, т. е. в ЭСО. В результате перекрывания волновых функций o-Ps и электронов атомных орбиталей, образующих стенки ЭСО, времена жизни o-Ps сильно укорачиваются по сравнению с аннигиляцией в вакууме и обычно составляют от 1.5 до 4.0 нс. Наблюдаемые времена жизни сильно зависят от размеров ЭСО: чем больше ЭСО, тем дольше время жизни позитрона в полимере. Спектр времен жизни — это набор экспериментальных характеристик времен i (нс) и соответствующих статистических весов или интенсивности Ii (%). Предполагается, что интенсивность позитрониевой компоненты зависит от концентрации ЭСО.
Источником позитронов обычно служит изотоп 22Na (период полураспада 2.6 года). Образующиеся позитроны имеют энергию от 0−0.5 МэВ с максимумом распределения 0.2 МэВ и длину пробега в обычных полимерах? 1 мм. В веществе позитроны быстро термализуются, и все последующие процессы протекают с участием частиц с тепловыми энергиями.
Экспериментальная установка для измерения времен аннигиляции позитронов, приведена на рис. 1. Она состоит из источника позитронов, помещенного между двумя образцами исследуемого полимера. Времена жизни измеряются электронной системой, работающей по принципу преобразования интервалов времени в амплитуду электрических импульсов. Схема регистрирует время между двумя событиями: появлением первичных г-квантов от источника, и квантов, сопровождающих аннигиляцию позитронов. После регистрации 105−107 таких событий (т.е. зарегистрированных фотоумножителем г-квантов) строят экспериментальную кривую распределения времен жизни позитронов y (t), показывающую число событий y в зависимости от измеренного времени t.
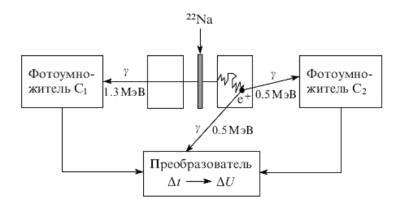
Рис. 1. Схема установки для измерения спектра времен аннигиляции позитронов Первичные экспериментальные зависимости y (t) могут интерпретироваться в рамках анализа, основанного на дискретной или непрерывной модели.
Предложено полуэмпирическое выражение, связывающее время жизни o-Ps (ф3) со средним размером радиуса (R3) сферического ЭСО в полимере (формула Тао-Элдрупа):
В предположении сферической геометрии по известному радиусу находят объем ЭСО (хf) или распределение по объемам.
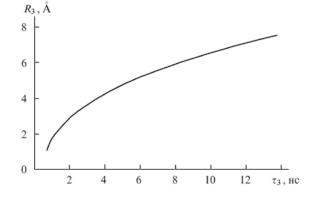
Рис. 2. Связь времени жизни позитрония и среднего радиуса ЭСО в полимерах согласно формуле Тао-Элдрупа Большие размеры элементов свободного объема были обнаружены в полимерных сорбентах и органических кластерах. Этим методом было найдено, что в сверхсшитых полистирольных сорбентах существуют полости с радиусом пор 14 A, на долю которых приходится около 20% от общего числа элементов свободного объема в материале.
Т.о., метод аннигиляции позитронов дает микроскопическое описание свободного объема в терминах средних радиусов ЭСО и соответствующих объемов хf. Чтобы связать эти величины с макросокопическими параметрами полимеров, необходимо узнать средние концентрации элементов свободного объема N (см-3), после чего можно оценить долю свободного объема как произведение хfN.
Традиционный метод аннигиляции позитронов направлен на изучение свободного объема внутри полимерной матрицы. Однако существует множество объектов, для которых важны не объемные, а поверхностные свойства полимеров (мембраны), покрытия на поверхности различных материалов и т. д. Такие свойства полимерных слоев, как температура стеклования, плотность, подвижность цепей и другие могут сильно отличаться внутри полимерной матрице и на границе раздела фаз.
Используемые в традиционной методике позитроны со средней энергией 200 кэВ имеют тормозной путь 1 мм. Это значение на много порядков больше, чем характеристическая толщина поверхностных слоев, поэтому времена жизни позитронов в подобных экспериментах несут информацию о свободном объеме в матрице. Для изучения свободного объема в приповерхностном слое, необходимо чтобы энергия пучка позитронов была существенно ниже указанного значения (позитроны не должны проникать в глубь объема образца и аннигилировать в приповерхностных слоях). Для этого используются монохроматические позитронные пучки малой энергии с существенно меньшей (0.2−20 кэВ) контролируемой энергией позитронов, что дает возможность зондирования свободного объема в тонких слоях полимеров (несколько нанометров) и построения профиля свободного объема в них.
Перечисленные выше методы изучения свободного объема в полимерах являются зондовыми, т. е. основаны на том, что в полимер вводят некоторые молекулы-зонды и следят за его поведением, на основании чего делают выводы о структуре свободного объема. Эти методы различаются природой и размерами выбранных зондов. Так, в методе аннигиляции позитронов для исследования полимеров зондом служит электрон-позитронная пара — атом позитрония. В методе обращенной газовой хроматографии в качестве зондов выступают ряды структурно-родственных соединений. Широкое применение нашел и метод ЯМР 129Xe, в котором единственный зонд — атом 129Xe — «обследует» свободный объем в разных полимерах.
Кроме экспериментальных методов изучения свободного объема в полимерах в последние годы все большее значение приобретают методы компьютерного моделирования наноструктуры полимеров с целью предсказания свойств полимерного материала (коэффициентов диффузии, растворимости и т. д.). При анализе данных компьютерного моделирования результаты зондовых методов используют для подтверждения достоверности расчетов. Вместе с тем, компьютерное моделирование открывает новые дополнительные возможности исследования свободного объема в полимерах, в основном принципиально недоступные зондовым методам его изучения: визуализация свободного объема; анализ связности свободного объема (размер кластеров, замкнутая или открытая пористость); построение распределения по размерам ЭСО; изучение мобильности (динамики) элементов свободного объема в полимерах.
В настоящее время для моделирования свободного объема широко применяются методы Монте-Карло, молекулярной механики, а также теория переходного состояния и родственные подходы.
Знание ряда термодинамических параметров и других физико-химических величин, полученных зондовыми методами для полимеров необходимо для определения потенциальной области их практического применения. Так, например, перфторированные полимеры обладают высокой газопроницаемостью и большим свободным объемом, повышенной химической устойчивостью (способность осуществлять разделение агрессивных сред), а также высокой термической стабильностью. К настоящему времени в материаловедении уже нашли применение фторированные полимеры Hyflon (статистический сополимер 2,2,4-трифтор-5-трифторметокси-1,3-диоксалана и тетрафторэтилена) и Cytop, структура которых представлена в таблице 1.

Таблица 1. Структура некоторых аморфных перфторированных полимеров Hyflon AD80X (80 мол.% диоксалана) Cytop.
Эти перфторированные полимеры характеризуются пониженными коэффициентами растворимости газообразных углеводородов, они нерастворимы в обычных органических растворителях, не набухают и не разрушаются при контакте с нефтепродуктами. Эти качества могут иметь важное значение при использовании этих полимеров в качестве материала газоразделительных мембран: низкая растворимость обычных органических соединений в перфторированных полимерах проявляется в пониженной способности к пластификации, а именно этим явлением обусловлено значительное ухудшение селективности газоразделительных и первапорационных мембран.
Природа большого свободного объема в аморфных перфторированных полимерах связана с высокой жесткостью цепей в этих полимерах, что затрудняет их плотную упаковку цепей при условии слабых межцепных взаимодействий.
Уже сейчас в литературе имеются данные по эффективности мембран на основе аморфного тефлона AF 2400 для проведения органоселективной первапорации (разделения смесей хлорпроизводных метана и очистки содержащих их сточных вод). Объектами такого разделения является область нефтехимии и химии тяжелого органического синтеза. Однако внедрение первапорации для разделения многочисленных азеотропных смесей ограничивается растворимостью существующих первапорационных мембран в разделяемых органических смесях и низкой проницаемостью.
Существенной проблемой является весьма ограниченный ассортимент доступных перфторированных полимеров в качестве мембранных материалов.
Диффузия газов в полимерах
Механизм диффузии газов и жидкостей близок к механизму их течения и состоит в последовательных периодически повторяющихся перескоках диффундирующих молекул из одного положения равновесия в другое, что возможно при наличии свободного объема. Чтобы произошел элементарный акт перескока диффундирующей молекулы, необходимы:
- — наличие по соседству с ней «дырки» нужного размера;
- — достаточной энергии для разрушения связи между молекулами полимера, т. е. энергии активации диффузионного процесса.
Количественно скорость диффузии оценивают по величине коэффициента диффузии, который представляет собой коэффициент пропорциональности в первом законе Фика:
Коэффициент диффузии выражается в см2/с, а в единицах СИ м2/с.
Но на основании первого закона Фика рассчитать коэффициент диффузии нельзя, так как этот закон справедлив только для стационарного потока. В общем случае имеют дело с нестационарным потоком, для которого градиент концентрации не постоянен. При этом справедлив второй закон Фика:
В настоящее время особое место занимают исследования сорбционной и десорбционной способности в полимерах сложной архитектуры (дендримеры, сверхразветвленные полимеры, звезды и т. д.) в отношении «гостевых» молекул. Сложность этих явлений, например, в случае разветвленных полимеров заключается в том, что сорбат локализуется во внутренних частях макромолекул (в сердцевине), а фактором, контролирующим кинетику сорбции (десорбции) служит оболочка из функциональных групп, диффузионная проницаемость которой зависит от природы межмолекулярных взаимодействий с участием молекул среды. Чрезвычайная важность исследования закономерностей процесса сорбция-десорбция заключается в том, что на нем основано уникальное свойство разветвленных макромолекул служить в качестве наноконтейнеров нерастворимых сорбатов и наноразмерных частиц в процессах фазопереноса, транспортировки фармакологических препаратов в организм человека.
Наиболее распространенными методами определения коэффициента диффузии D являются сорбционно-десорбционный и метод Дейнеса-Баррера.
Метод Дейнеса-Баррера заключается в следующем. Используют замкнутую ячейку, состоящую из двух металлических камер, которые разделяют испытуемой мембраной из полимерного материала. С одной стороны создается давление P исследуемого газа, а с другой стороны — вакуум. При соприкосновении газа с одной из сторон мембраны его появление с другой стороны наблюдается через определенный промежуток времени. После чего, продиффундировавшего внутрь ячейки через пленку, от времени.
Сорбционно-десорбционный метод. Этот метод широко используется для изучения диффузионной проницаемости полимеров благодаря своей простоте. Сущность метода состоит в изучении кинетики сорбции газа или пара образцом полимера в изобарно-изотермических условиях.
Если образец, сорбирующий газ, имеет форму пластины (пленки) толщиной l, то при граничных условиях 0 < x < l и 0 < c < cравн решение второго закона Фика имеет вид:
где Mt — количество сорбированного или десорбированного вещества к моменту времени t; M? — равновесное количество сорбированного вещества.
По полученным данным строят график зависимости Мt / М? = f (t½). Для систем, подчиняющихся второму закону Фика («фиковские системы») этот график имеет вид Г-образной кривой с начальным прямолинейным участком в области Мt / М ?, < 0.6 (рис. 4, кривая 1).
Г-образная зависимость наблюдается в том случае, когда D является функцией только концентрации и не зависит от времени. Во многих случаях наблюдается аномальная «нефиковская» диффузия, проявляющаяся в ином характере зависимости.
Мt / М? = f (t½),.
которая имеет вид S-образных или двухступенчатых кривых (рис. 4, кривая 3). Отклонения от второго закона Фика обычно связаны с изменением конформации макромолекул, а также с перемещениями структурных элементов полимера, происходящими в результате взаимодействия его макромолекул с молекулами диффундирующего газа. Оба эти процесса имеют релаксационный характер. Если время релаксации невелико, что наблюдается у полимеров, находящихся в эластическом состоянии, то конформационные и структурные изменения происходят достаточно быстро. В этом случае D зависит только от скорости перемещения молекул газа. При Т < Тс подвижность сегментов мала, и скорость конформационных и структурных превращений может оказаться ниже скорости диффузии газа. Суммарный эффект переноса определяется скоростью первых процессов, и D является функцией времени.
Mt/M?
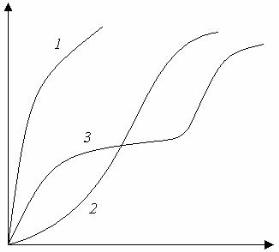
Рис. 4. Кинетические кривые сорбции: 1 — фиковская диффузия; 2 и 3 — нефиковская диффузия t½.
4.3 Механические методы
Динамический механический анализ (ДМА). Этот метод анализа применяется для исследований зависимости механических и вязкоупругих свойств материалов от температуры. В результате исследований можно определить модуль Юнга и модуль сдвига, данные о структуре и морфологии полимеров, релаксационные характеристики и вязкоупругие свойства, дефекты материалов, также проанализировать разрушение полимеров. Использование метода ДМА в настоящее время в значительной мере помогло оптимизировать и сделать максимально эффективным процесс переработки полимеров за счет корреляции между технологическими условиями и молекулярной структурой полимера.
Этот метод является чувствительным методом определения механического отклика полимерного материала при помощи слежения за изменением свойств в зависимости от температуры и частоты приложенного синусоидального напряжения. Уникальность современного оборудования заключается:
- — в одновременном измерении нагрузки и смещения, что позволяет получать очень точные значения модуля упругости;
- — возможности измерения в широком диапазоне амплитуд нагрузки (от 1 мН до 40 Н), что позволяет исследовать как мягкие, так и жесткие материалы и
в широком диапазоне частот нагружения (от 0.001 Гц до 1000 Гц).
В этом методе к образцу прикладывается усилие и измеряется амплитуда и фаза результирующего смещения. ДМА использует линейный исполнительный механизм, в котором приложенное к образцу усилие (напряжение) рассчитывается из сигнала, подаваемого на электромагнитные катушки двигателя. Синусоидальное напряжение, которое прикладывается к образцу, создает синусоидальную деформацию или смещение. Это прикладываемое напряжение (усилие) выбирается таким малым, чтобы не изменить анализируемый материал. Измеряя как амплитуду деформации в пике синусоидной волны, так и разность фаз между синусоидальными волнами напряжения и деформации, можно рассчитать такие величины, как модуль, вязкость и затухание. Когда отклик материала на приложенные колебания является совершенно упругим, входной сигнал находится в фазе с выходным, то наблюдается фазовое запаздывание д = 0?, если вязкий отклик дает расхождение фаз, то д = 90?. Вязкоупругие материалы находятся между этими двумя предельными случаями, т. е. 0? > д < 90?.
Данный метод разделяет динамический отклик материалов на две различные части — упругую часть D' и вязкостную компоненту D''. Комплексный модуль D* определяется как мгновенное отношение фазового или упругого отклика D' (который пропорционален восстановимой или запасенной энергии) к вязкостному отклику D'' (который пропорционален невосстановимой или рассеянной энергии).
D* = D' + iD''.
Датчик силы и смещения.

Рис. 5. Динамический-механический анализатор Мотор платы.
Тангенс угла механических потерь tgд еще один важный параметр для сравнения вязкоупругих свойств различных материалов:
tgд = D''/D',
где tgд (фактор затухания) является отношением диссипированной и запасенной энергии. Наличие максимума зависимости tgд от температуры является признаком релаксационного перехода, а также фазовых превращений. Это обусловлено тем, что в процессах фазовых и релаксационных переходов из одного состояния в другое совершается перестройка структуры, при которой суммируются потери, типичные для обоих состояний. Динамический метод очень удобен для оценки процесса структурирования и вулканизации полимеров, чему соответствует расширение зоны высокоэластичности и снижение потерь в области низких частот.
Данный метод завоевал большую популярность вследствие его быстроты и высокой точности, а так же способности сканировать материалы в широкой области температур и частот.
Кроме этого, данный метод в настоящее время используют как альтернативный подход к изучению разветвленных полимеров в режиме вязкого течения в сочетании с реометрическими измерениями, с помощью которых можно измерить ползучесть полимерного материала. Это связано с ограниченностью стандартных методов по отношению к полимерам сложной архитектуры, поскольку их релаксационные свойства обусловлены не межмолекулярными взаимодействиями типа зацеплений (как в случае линейных полимеров), а внутримолекулярными перестройками, т. е. связаны непосредственно с их топологической структурой. Так группой исследователей (Врана К. и др.) на примере сополимеров разветвленного строения на основе этилена, пропилена и винилнорборнена было показано, что, сочетая два перечисленные метода с помощью специальных диаграмм (Диаграмма Ван Гурпа-Пальмена) можно проследить не только влияние структуры цепи на текучесть полимеров, но и определить степень их разветвленности.
На рис. 6. представлены зависимости модуля сдвига сополимеров, полученных на основе трис-(пентафторфенил)германа и бис-(пентафторфенил)германа различной степени разветвленности. Наиболее характерным является увеличение значения модуля с температурой, что может быть связано с увеличением взаимодействия макромолекул вследствие взаимопроникновения фрагментов наружной сферы. По-видимому, размораживание конформационной подвижности этой сферы ответственно за высокотемпературный релаксационный переход, который может быть принят за температуру стеклования. Из рисунка видно, что Тс сополимеров возрастает до 252 С (максимумы на кривых) при увеличении содержания бис-(пентафторфенил)германа до 50.7%. Для разветвленного перфторированного полифениленгермана, полимера полученного из трис-(пентафторфенил)германа путем активированной поликонденсации Тс=163 С. Эффект увеличения Тс может быть обусловлен межи внутримолекулярной сшивкой дендронов, и как следствие, ограничением вращения вокруг связей Ge-C.
Прочностные характеристики полимеров. Широкое использование полимерных материалов определяется также и их ценными механическими свойствами и высокой прочностью в сочетании со способностью к большим обратимым деформациям. Создание новых полимерных материалов с заранее заданными функциональными свойствами является одной из главных проблем современной полимерной науки. Выполнение этой задачи требует знания фундаментальных закономерностей, связывающих физико-химические свойства полимерных материалов с их молекулярной структурой, а также разработки на этой основе методов управления свойствами композиционных систем. Хорошо известно, что смешение полимеров позволяет создавать материалы, характеристики которых являются промежуточными между свойствами исходных компонентов. Примером таких смешивающихся полимерных систем являются композиционные материалы.
Рассмотрим древесно-полимерные композиции (ДПК), которые в последнее время нашли широкое применение в строительстве в качестве декинга (террасная доска). Потребность рынка в ДПК обусловлена некоторыми недостатками природного дерева, такими как высокое водопоглощение, большая микробная деструкция и малая долговечность. В связи с чем, ДПК уже нашли применение не только для изготовления террасных досок, но и для изготовления фундаментов, дощатых тротуаров, сайдинга, паллетов, кровельной черепицы. Кроме того, из композитных материалов с длинноволокнистой целлюлозой изготавливают автомобильные изделия (внутренние панели, запасные покрышки и т. д.). ДПК могут быть получены из полимеров термопластов любого типа в сочетании с наполнителями (целлюлозное волокно) и другими компонентами.
Древесно-полимерные композиты изготавливают из полимера, наполненного целлюлозным волокном и другими компонентами путем экструзии или формования. Основное требование к полимеру — термопласты, с температурой переработки ниже 200 С, что обусловлено невысокой термостойкостью древесины. Важным критерием при производстве ДПК являются характеристики текучести расплава, что связано с режимами переработки при производстве и может привести к появлению дефектов на поверхности экструдированных профилей, шероховатости поверхности. Такие проблемы могут возникнуть из-за плохой текучести состава при данных температурах переработки, и она может быть заранее выявлена путем учета реологии наполненной композиции или самого полимера на стадии выбора. Т.о. определяющую роль при создание рецептур ДПК играют реологические особенности выбранных термопластичных полимеров (скорость сдвига, сдвиговое напряжение, динамическая вязкость, предельная вязкость).
Наиболее изученными термопластами являются полиэтилен, полипропилен, поливинилхлорид, сополимеры стирола с акрилонитрилом и бутадиеном, полиамид и т. д., использование композиций с поливиниловым спиртом и полиакрилатом привело к созданию биоразлогаемых ДПК. Для улучшения совместимости полимеров и наполнителей при получении ДПК используют связующие агенты, которые могут химически связываться и с целлюлозным волокном и (или) сополимеров, что способствует образованию гомогенной системы.
Механические свойства ДПК в значительной степени определяются природой наполнителя и полимера. Рассмотрим основные механические характеристики, связанные с областью применения ДПК:
- — прочность на изгиб, которая определяет разрушающую нагрузку (предельная нагрузка), до значения которой может эксплуатироваться данная композиция;
- — модуль упругости и прогиб при изгибе. Эта характеристика тесно связана
с разрушающей нагрузкой, и ее величина определяется требованиями строительных норм. Прогиб при однородно распределенной нагрузке определяется нагрузкой, шириной доски, модулем упругости при изгибе, моментом инерции (мера прочности и жесткости доски, или фактор жесткости) и пролетом между опорами.
Кроме перечисленных механических свойств важными критериями являются термическое расширение-сжатие, усадка, сопротивление скольжению, водопоглощение, микробная деструкция, горючесть, окисление и выцветание.
В качестве минеральных наполнителей используют карбонат кальция, кремнезем, тальк, что повышает жесткость наполненного продукта и придает полимеру более высокую огнестойкость, кроме того, их введение обычно улучшает как прочность при изгибе (обычно на 10−20%), так и модуль упругости при изгибе (на 200−400%) наполненных ДПК. Использование целлюлозы в качестве наполнителя в термопластичных композициях ранее (1970;х гг.) было затруднительно, поскольку она плохо распределяется в полимерных рецептурах при формовании и компаундировании. В связи с чем, рядом исследователей был получен композитный материал, включающий термопластичный ПВХ и целлюлозное волокно в виде древесной массы или хлопкового пуха (Патент США № 3 943 079). Данная проблема была так же решена при использовании целлюлозного волокна, полученного от таких материалов как древесные опилки, льна, соломы и пшеницы, и связующего смешанного с наполнителем (Патент США № 6 939 496). Патентный поиск показал, что для улучшения механических свойств ДПК, при производстве удаляют влагу из целлюлозных волокон (древесной массы, отходов бумаги и т. д.) перед их смешением с термопластичными полимерами (полипропилен, полиэтилен, поливинилхлорид и т. д.), что приводит к получению литьевых изделий без полостей и пузырей, и улучшает физические свойства формованных продуктов (Патент США № 4 687 793).
4.4 Электрические методы
В основе метода лежат исследования температурно-частотной зависимости тангенса угла диэлектрических потерь (tg) и диэлектрической проницаемости (). Диэлектрическая проницаемость определяется отношением емкости электрического конденсатора, заполненного этим веществом, к емкости того же конденсатора в вакууме при определенной частоте внешнего поля. Эта величина связана с поляризацией, т. е. с возникновением определенного электрического момента в единице объема диэлектрика при внесении его в электрическое поле. Часть энергии электрического поля, которая необратимо рассеивается в диэлектрике в форме теплоты (диссипация энергии) называется диэлектрическими потерями. Угол, который определяется сдвигом фаз между векторами приложенного к диэлектрику электрического поля и поляризации, возникающей под действием этого поля называется углом диэлектрическких потерь —, а его тангенс — тангенсом угла диэлектрических потерь.
Т.к. диэлектрические свойства полимеров зависят от химического строения
и структуры повторяющегося звена, строения макроцепей и способа их укладки, этот метод используется для изучения молекулярной структуры и теплового движения в полимерах.
Особенностью полимеров является независимое движение отрезков цепи, состоящих из большого количества сегментов. Кроме движения сегментов в полимере возможно движение более мелких и более подвижных кинетических единиц (боковые цепи, например полярные заместители). Время релаксации ориентационного момента таких групп меньше времени релаксации сегментов главной цепи, поэтому они могут сохранять подвижность при более низких температурах, при которых сегменты ее уже не проявляют. Если полимер, содержащий полярные группы поместить в электрическое поле, при определенных соотношениях времен релаксации и частоты поля будет происходить ориентация сегментов и более мелких кинетических единиц, что обуславливает некоторые значения диэлектрической проницаемости и диэлектрических потерь. В данном случае важное значение имеет не только полярность групп в полимере, но и способ их вхождения в мономерное звено. Т. е. химическое строение повторяющегося звена оказывает существенное влияние на внутрии межмолекулярные взаимодействия, т. е. на подвижность звеньев и время релаксации. Чем сильнее внутрии межмолекулярные взаимодействия, тем подвижнее звенья, тем выше температура, при которой наблюдается максимум tg, и тем больше время релаксации.
На диэлектрические свойства также влияют:
- — изомерия повторяющегося звена (например способ присоединения эфирного кислорода в полиметилметакрилате и поливинилацетате);
- — наличие в макромолекуле участков синдиотактического или изотактического строения. Их протяженность и количественное соотношение существенно влияют на подвижность сегментов и групп, а следовательно, и на диэлектрические характеристики полимеров.
Кроме перечисленных факторов на диэлектрические свойства также влияет ориентационная вытяжка макромолекул. Растяжение полимера может приводить как к увеличению, так и к уменьшению времени релаксации диэлектрических потерь, связанных с ориентационными поворотами полярных звеньев макромолекулы в условиях, когда возможно сегментальное движение (высокоэластическое состояние) в зависимости от того, происходит ли при растяжении уплотнение или разрыхление упаковки макромолекул.
5. Свойства полимеров в монослоях ленгмюра и в тонких пленках
Некоторые характеристики полимерных пленок и монослоев (поверхностное натяжение, угол смачивания, топография поверхности, самоорганизация на границе раздела фаз вода-воздух) являются важными параметрами, например, для контроля процессов очистки поверхностей, загрязненных нефтепродуктами, при исследовании свойств биоматериалов (линзы, импланты) фармацевтических препаратов (порошки, таблетки, капсулы).
5.1 Изучение коллоидно-химических свойств дифильных макромолекул в монослоях и пленках Ленгмюра-Блоджетт
В настоящее время в развитии полимерной химии большое внимание уделяется исследованиям дифильных полимеров, которые в своем составе имеют блоки различной природы. Появление у них новых интересных свойств связано со способностью макромолекул к самоорганизации в растворе и в массе. Определяющую роль при самоорганизации в тонких пленках играет несовместимость ковалентно связанных блоков. Основными факторами, которые будут определять морфологию поверхности пленок сополимера являются соотношение длин различных блоков и характер взаимодействия между ними. Для таких блок-сополимеров преимущественно характерны такие структуры как сферы, цилиндры, ламели. Кроме того, гидрофобные и гидрофильные группы в составе макромолекул блок-сополимеров придают молекуле амфифильные свойства, что является необходимым условием для образования устойчивых мономолекулярных пленок на границе раздела фаз. Причем, характер процессов, протекающих при образовании пленок на границе раздела фаз вода-воздух, в первую очередь зависит от гидрофильно-гидрофобного баланса в молекулах блок-сополимеров.
Значительные успехи в области молекулярной архитектуры, достигнутые в последние два десятилетия, во многом обусловлены использованием такого метода получения пленок дифильных соединений как метод Ленгмюра-Блоджетт. Этот метод является удобным и эффективным способом диспергирования вещества до молекулярного уровня, исследования поверхностных свойств и формирования ультратонких монои мультимолекулярных пленок, в том числе аналогов липидных мембран. Кроме того, технология Ленгмюра-Блоджетт позволяет достаточно просто изменять свойства поверхности и формировать качественные пленочные покрытия за счет точного контроля толщины пленки (количества наносимых слоев) в процессе выделения, однородности покрытия, низкой шероховатости и высокой (при подборе адекватных условий) адгезии пленки к поверхности. Свойства пленок можно также легко варьировать, изменяя структуру полярного блока амфифильной макромолекулы, состав монослоя (двухи многокомпоненные смеси молекул), а также условия выделения (состав субфазы и поверхностное давление). Т. е. управляя размерами и формой макромолекул, можно придавать материалам совершенно новые функциональные качества, резко отличающиеся от имеющихся у обычных полимеров.
Изотермы поверхностного давления и перенос монослоя. Формирование упорядоченого монослоя на поверхности субфазы происходит следующим образом. Определенный объем раствора исследуемого вещества в легколетучем растворителе наносится на поверхность субфазы. После испарения растворителя на поверхности воды образуется мономолекулярная пленка, молекулы в которой расположены хаотически. При постоянной температуре T состояние монослоя описывается изотермой сжатия р-А, отражающей соотношение между величиной поверхностного давления барьера р и удельной молекулярной площадью А (Рис. 1, а).
а б Рис. 1. Изотерма Ленгмюра (а) и поведение макромолекул на границе раздела фаз вода-воздух (б) С помощью подвижного барьера монослой сжимается до получения сплошной пленки с плотной упаковкой молекул, в которой удельная молекулярная площадь А приблизительно равна площади поперечного сечения молекулы, а углеводородные радикалы ориентированы почти вертикально (рис. 1, б). Линейные участки на зависимости р-А, отвечающие сжатию монослоя в различных фазовых состояниях, характеризуются величиной А0 — площадью приходящейся на молекулу в монослое, полученной экстраполяцией линейного участка на ось А (р = 0 мН/м). Фазовое состояние локализованного на границе раздела «субфаза-газ» монослоя амфифильного вещества определяется адгезионно-когезионным балансом сил в системе «субфаза-монослой» и зависит от природы вещества и строения его молекул, температуры T и состава субфазы. Выделяют газообразные G, жидкие L1, жидко-кристаллические L2 и твердо-кристаллические S монослои (рис. 1, б).
С помощью микроскопа Брюстера, который представляет собой видеокамеру с лазерным приводом, можно наблюдать в режиме реального времени з, а пленками Ленгмюра на границе раздела фаз вода-воздух или на подложке из диэлектрика.

Сформированный монослой, состоящий из плотноупакованных молекул, переносится на движущуюся вниз-вверх через поверхность воды твердую подложку. В зависимости от типа поверхности подложки (гидрофильная или гидрофобная) и последовательности пересечения подложкой поверхности субфазы с монослоем и без монослоя, можно получать пленки Легнмюра-Блоджетт (ПЛБ) симметричной (Y) или асимметричной (X, Z) структуры (рис. 3).
Технология получения тонких пленок Ленгмюра-Блоджетт, с помощью которой можно легко управлять размерами и формой макрообъектов в последнее время активно развивается, поскольку с помощью нее можно придавать полимерным материалам новые функциональные свойства. Основная проблема при создании наноструктурированных полимерных пленок связана с подбором оптимальных условий формирования пленок, которые в определенной степени зависят от молекулярной массы гидрофильного и гидрофобного блоков, от степени разветвленности дендритного фрагмента в гибридных блок-сополимерах, от природы линейного блока. ПЛБ представляет собой мультислой — принципиально новый объект современной физики, и потому любые его свойства (оптические, электрические, акустические и т. д.) совершенно необычны. Даже простейшие структуры, составленные из одинаковых монослоев, имеют ряд уникальных особенностей, не говоря уже о специально построенных молекулярных ансамблях.
Значение поверхностного давления, при котором проводится перенос монослоя на подложку, определяется по изотерме сжатия данного амфифильного сополимера и соответствует состоянию с плотной упаковкой молекул в монослое. В процессе переноса давление p поддерживается постоянным за счет сокращения площади монослоя движущимися барьерами. Критерием степени покрытия подложки монослоем, является коэффициент переноса k, который определяется по формуле:
Рис. 3. Пленки Ленгмюра-Блоджетт Для получения однородной по толщине пленки Ленгмюра-Блоджетт, поверхность подложки должна иметь шероховатость Rz? 50 нм. Из изотерм поверхностного давления можно извлечь информацию как об универсальных эффектах межмолекулярного взаимодействия в монослое, так и о специфике поведения сложной амфифильной молекулы при изменении поверхностного давления (ее переориентации, конформационных перстройках и т. д.). Как правило, разрушение монослоя таких молекул происходит при высоких поверхностных давлениях (до 30 мН/м). Низкие значения поверхностного давления, возможно, связаны с ассоциацией амфифильных молекул друг с другом (образование димеров, триммеров и т. д.), что приводит к уменьшению числа частиц, образующих двумерный газ, с образованием макроскопических островков твердой фазы при малых давлениях. Кроме того, при интерпритации полученных р-А изотерм необходимо учитывать стерические отталкивания и дисперсионные притяжения между углеводородными цепочками, а также кулоновские и диполь-дипольные силы взаимодействия между полярными группами. Также следует помнить, что амфифильные молекулы могут сильно взаимодействовать с водой, образуя водородные связи.
Рассмотрим изотерму поверхностного давления блок-сополимера поли-(N-винилпирролидон-2,2,3,3-тетрафторпропилметакрилат) (Рис. 4). Участок полученной изотермы, при давлении близком к нулю, соответствует области двумерного газа, в котором молекулы блок-сополимера лежат на поверхности раздела фаз и не контактируют друг с другом. При дальнейшем сжатии пленки молекулы начинают контактировать в области двумерной жидкости, и наблюдается первый рост поверхностного давления, при достаточно большом уменьшении площади пленки.
В области фазового перехода на изотерме при практически постоянном давлении присутствует линейный участок (плато), где макромолекулы начинают перестраиваться и менять форму: гидрофильные группы погружаются в воду, а гидрофобные остаются на поверхности и выстраиваются перпендикулярно границе раздела фаз. При дальнейшем сжатии барьера происходит второе, резкое увеличение поверхностного давления при малом уменьшении площади пленки, что дает основание говорить о формировании «двумерного твердого тела» и окончании перестроения поверхностных групп.
В этой области изотермы пленка находится в максимально сжатом состоянии, и дальнейшее ее сжатие приводит к разрушению монослоя.
В некоторых случаях на изотерах поверхностного давления может наблюдаться негоризонтальность области «плато», что видимо, связано с уходом молекул с поверхности в объем водной фазы. Кроме того, несовпадение изотерм сжатия и расширения свидетельствует о некоторой агрегации блоков макромолекул в процессе сжатия.
5.2 Поверхностные свойства пленок
Явления, наблюдаемые на поверхности твердых тел, определяются физико-химическими взаимодействиями фаз на поверхности раздела. В последнее время наиболее распространенным методом изучения свойств поверхности является смачивание, при котором интенсивность этих взаимодействий характеризуется, прежде всего, величиной краевого угла между поверхностями жидкости и твердого тела на границе с окружающей средой.
В зависимости от числа фаз, участвующих в смачивании, различают два основных случая. Смачивание при полном погружении твердого тела в жидкость (иммерсионное смачивание), в котором участвуют только две фазы — жидкость и твердое тело, и контактное смачивание, в котором, наряду с жидкостью, с твердым телом контактирует третья фаза — газ или другая жидкость. В первом случае интенсивность взаимодействий на поверхности твердых тел характеризуется теплотой смачивания. Во втором — величиной краевого угла (угла между поверхностями жидкостями и твердого тела на границе с окружающей средой).
Основной характеристикой состояния поверхности твердых тел служит поверхностное натяжение у, характеризующее нескомпенсированность межмолекулярного взаимодействия на границе раздела фаз и обуславливающее избыток поверхностной энергии. Оно определятся, как работа обратимого изотермического процесса образования единицы площади поверхности раздела фаз, при условии, что термодинамические параметры состояния системы (температура, давление, химические потенциалы компонентов) не изменяются. Согласно подходу Фоукса величина поверхностного натяжения складывается из 3-х составляющих: полярной (индукционной) (p), дисперсионной (d) и ориентационной (0). d — эффективное поверхностное натяжение, обусловленное межмолекулярным взаимодействием жидкости с твердой поверхностью, pповерхностное натяжение, обусловленное индукционным (полярным) взаимодействием на границе жидкость — твердое тело. Ориентационная составляющая мала и ее обычно не рассматривают.
Дисперсионные и полярные составляющие поверхностного натяжения можно определить методом Зисмана, согласно которому вклады p Т — Г и d Т — Г определяют по изменению и0 в системе твёрдое тело — вода (ж1) — углеводород (ж2). Для этой системы уравнение Юнга будет иметь следующий вид:
Т-Ж1= Т-Ж2+ Ж1-Ж2 cos и0, (2).
где — Т-Ж1, Т-Ж2 и Ж1-Ж2 — поверхностные натяжения на границах раздела фаз: твёрдое тело — жидкость 1, твёрдое тело — жидкость 2, жидкость 1 — жидкость 2 соответственно. Тогда уравнения Фоукса запишутся в виде:
Т-Ж2= Т-Г + Ж2-Г — 2(dТ-Г х dЖ2-Г)½ — 2(РТ-Г х РЖ2-Г)½, (3).
Т-Ж1= Т-Г + Ж1-Г — 2(dТ-Г х dЖ1-Г)½ — 2(РТ-Г х РЖ1-Г)½, (4).
где Т-Ж1, Т-Ж2, Ж1-Ж2 — поверхностные натяжения на границах: твердое тело — вода, твердое тело — углеводород, вода — углеводород;
Ж1-Г, dЖ1-Г, РЖ1-Г, Ж2-Г, dЖ2-Г, РЖ2-Г, Т-Г, dТ-Г, РТ-Г — поверхностные натяжения и их дисперсионные и полярные составляющие чистых веществ на границе с воздухом.
Вычитая уравнения (3) — (4) с учетом того, что для углеводородов РЖ2-Г= 0 (Ж2-Г = dЖ2-Г) получим:
Т-Ж2- Т-Ж1= Ж2-Г — Ж1-Г +2(dТ-Г)½ [(dЖ1-Г)½ -(Ж2-Г)½].
+2(РТ-Г х РЖ1-Г)½, (5).
учитывая уравнение Юнга (2) окончательно получаем:
Ж1-Г — Ж2-Г + Ж1-Ж2 cos 0 = 2(dТ-Г)½ [(dЖ1-Г)½ — (Ж2-Г)½] +.
+2(РТ-Г х РЖ1-Г)½, (6).
где значения Ж1-Г, Ж2-Г, Ж1-Ж2, dЖ1-Г, Ж2-Г, РЖ1-Г — справочные величины.
Зависимость в координатах: (Ж1 — Ж2 +.
Ж1-Ж2 cos) от 2 [(dЖ1-Г)½ ;
(Ж2-Г)½] описывается уравнением прямой типа.
y = x tg + b (рис. 5.).
Ж1-Г — Ж2-Г + Ж1-Ж2 cos.
2х [(dЖ1-Г)½ — (Ж2-Г)½].
Рис. 5. График зависимости для определения dТ-Г, РТ-Г Из полученного графика можно рассчитать dТ-Г, РТ-Г, Т-Г по следующим формулам:
Следует отметить, что такой подход справедлив только в том случае, когда вода вытесняет углеводород на поверхности твердого тела, то есть выполняется неравенство:
Т-Ж1 — Т-Ж2 — Ж1-Ж2 0 (10).
В качестве примера в таблицах 1, 2 приведены данные метода смачивания и расчеты поверхностного натяжение и его составляющих методом Зисманапленок блочного сополимера ПММА-ПФГ, полученных из различных растворителей (хлороформ, ТГФ).
Таблица 1. Средние значения краевых углов смачивания для пленок блочных сополимеров ПММА-ПФГ (пленка из хлороформа).
Состав. | ||||||
ППМА: ПФГ. | Вода/вода. | Вода/гексан. | Вода/гептан. | Вода/октан. | Вода/гексадекан. | |
(мас.%). | ||||||
ПММА. | 73.54. | 76.5. | 83.9. | 82.4. | 81.7. | |
99%-1%. | 71.7. | 80.1. | 84.5. | 82.6. | 83.7. | |
95%-5%. | 75.9. | 78.9. | 83.1. | 82.7. | 80.1. | |
90%-10%. | 72.1. | 79.4. | 81.3. | 81.8. | 79.8. | |
80%-20%. | 73.8. | 79.9. | 80.3. | 79.9. | 76.8. | |
Расчет полярной и дисперсионной составляющей для пленок блочного сополимера ПММА-20%ПФГ (хлороформ, тетрагидрофуран).
Таблица 2. Энергетические характеристики поверхности исследуемых пленок блочного сополимера ПММА-ПФГ (20 мас. % ПФГ), сформированных из хлороформа и ТГФ.
ПММА-ПФГ,. | Поверхностная энергия, мДж/м2. | и (H2O),. | ||||||
растворитель. | p | d | s | |||||
s | s | град. | ||||||
хлороформ. | 17.5. | 9.5. | ||||||
ТГФ. | 25.2. | 44.3. | 69.5. | |||||
Для пленок полимеров, когда возможны как дисперсионные, так и полярные взаимодействия, наиболее достоверные результаты при оценке свободной поверхностной энергии пленки полимера дает уравнение Оуэнса-Вэндта, которое связывает экспериментально определенный краевой угол смачивания и энергетические характеристики жидкости и твердого тела следующим образом: В данном методе используются тестовые жидкости различной природы, такие как вода, глицерин, диметилсульфоксид, гексан, додекан.
Данный метод широко применяется для пленок фторированных полимеров, и для блок-сополимеров на их основе, с преимущественным содержанием гидрофобного блока, где в основном преобладают дисперсионные силы, обусловленные возникновением водородных связей и диполь-дипольными взаимодействиями.
В настоящее время имеются полностью автоматизированные и контролируемые компьютером приборы (например, оптический тензиометр, рис. 3), которые в значительной мере упрощают работу исследователей. Они одновременно могут определять ряд параметров, таких как контактные углы смачивания, поверхностное и межфазное натяжение, свободную поверхностную энергию, пористость поверхности пленки и межфазную реологию.

Рис. 3. Оптический тензиометр В последнее время внимание исследователей направлено на получение структурированных поверхностей с заданной двумерной организацией на нано-или микромасштабе. Такие поверхности, обладающие интересными физическими и биологическими свойствами, могут быть использованы в качестве платформ для сборки более сложных функциональных наноструктур и наноустройств. В связи с чем, в настоящее время существует множество способов формирования наноструктурированных поверхностей, например, способ Ленгмюра-Блоджетт, получение путем осаждения модифицированных органическими молекулами наночастиц из их коллоидных растворов, а также подход, основанный на контролируемом взаимодействии (самоорганизации) молекул и/или наночастиц, выступающих в качестве «строительных блоков», такие поверхности могут служить в дальнейшем для создания более сложных структур. Во всех случаях незаменимым методом изучения природы поверхности пленок является атомно-силовая микроскопия.
5.3 Атомно-силовая микроскопия
Метод АСМ позволяет получать уникальную информацию о строении поверхности, определять деформационно-прочностные и релаксационные свойства индивидуальных макромолекул. Кроме того, с помощью этого метода можно проводить исследования по измерению энергии специфического взаимодействия между различными функциональными группами органических и биологических макромолекул. В последние годы появилась тенденция использования АСМ in situ в исследованиях плавления кристаллических и жидкокристаллических фаз.
В качестве примера на рис. 4, представлена топография поверхности пленки механической смеси полиметилметакрилата и перфторированного полифениленгермана (60%-40%). Из рисунка видно, что рельефы пленки механической смеси и пленки линейно-денритного блок-сополимера, для которого характерна равномерная гетерогенная поверхность, значительно отличаются. Топография пленки механической смеси обусловлена сегрегацией макромолекул разветвленного полимера в поверхностный слой пленки. В случае блок-сополимера аналогичному разделению компонентов мешает химическая связанность блоков.

А.

б.
Рис. 4. Топография поверхности пленок блок-сополимера ПММА — ПФГ (15 мас.% ПФГ) (а) и механической смеси ПММА-ПФГ (40% ПФГ) (б).
Разнообразие материалов, применяемых в различных новых технологиях, ставит задачу по созданию универсальных методов исследования, обладающих широким практическим применением, высокой разрешающей способностью и позволяющих при этом достоверно определять различные локальные свойства исследуемых объектов, в связи с чем, в настоящее время совершенствуется аппаратное решение ряда приборов для изучения поверхностных свойств, с целью расширения экспериментальных возможностей.
- 6. Практические работы
- 6.1 Работа 1. Изучение кинетики радикальной полимеризации бутилметакрилата в присутствии небольших количеств сверхразветвленного полимера на термографической установке
Теоретическая часть
Теоретическая часть подробно рассмотрена в главе 1 настоящего учебно-методического пособия.
Экспериментальная часть
Схема термографической установки показана на рис. 1.
ПК. | ||||||||||||||
ТА-5. | ||||||||||||||
Рис. 1. Схема термографической установки. | ||||||||||||||
1 — корпус ячейки, 2 — металлическая гильза, 3 — втулка из оргстекла, 4 — термометр сопротивления, 5 — тензометрический усилитель и электронный потенциометр, 6 — компьютер.
Изменение температуры в процессе полимеризации вызывает изменение сопротивления термометра сопротивления рабочей ячейки, в то время как сопротивление эталонной ячейки, остается постоянной. Регистрирующий и эталонный термометры сопротивления включаются в плечи моста, разбалланс с которого подается на усилитель ТА-5, затем сигнал с усилителя поступает на электронный потенциометр и далее на персональный компьютер (ПК). Применение установки такого типа позволяет регистрировать с большой точностью незначительные изменения температуры в процессе реакции порядка 10−4 градуса на миллиметр шкалы самописца, что позволяет регистрировать скорость полимеризации порядка 0.01% в минуту и выше.
Подготовка ячеек для изучения кинетики радикальной полимеризации до глубоких степеней превращения
- 1. Приготовить мономерные смеси для 4-х ячеек различного состава (ПБМА, и его смесь с сверхразветвленным перфторированным полифениленгерманом в количестве 0.01%, 0.05%, 1% и 5%). Объем мономерной смеси приготовить исходя из объема ячеек.
- 2. Заполнить ячейки мономерными смесями и дегазировать их трехкратным перемораживанием в вакууме. Ампулы отпаять.
Последовательность работы на термографической установке
- 1. Включить термостат, и после достижения температуры, при которой проводится полимеризация, прогреть ячейки 1−1.5 часа для установления в центре ячеек рабочей температуры.
- 2. Включить тензостанцию ТА-5 и дать ей прогреться в течении 15−20 минут.
- 3. Когда установка вышла на режим (данные по каждому из каналов не сильно отличаются), загружают ампулы с мономерными смесями, вставляя их в держатели образцов рабочих ячеек. В держатель образца эталонной ячейки вставляют эталонную ампулу (ПБМА). Помещают ампулы в соответствующие ячейки предварительно прогрев 3−5 минут при температуре опыта в стеклянных стаканах.
- 4. Показания прибора на компьютере регистрируются каждые 2 минуты, затем каждые 5 минут. Данные заносятся в таблицу (Таблица 1).
Таблица 1. Даные расчета кинетических данных по термографическим кривым.
Время ф,. | канал. | канал. | канал. | канал. | Т00. | Т01. | Т02. | Т03. | |
мин. | |||||||||
После чего, для каждого из образцов строят зависимость Т f (). Далее, используя программу в XL для построения кинетических кривых, заносят полученные данные и строят зависимость конверсии от времени и скорости полимеризации от времени:
Рис. 2. Зависимость степени превращения Рис. 3. Зависимость скорости превращения ММА от времени в присутствии различного ММА от времени в присутствии различного количества ПФГ. количества ПФГ.
После окончания полимеризации ампулы извлекают из термографической установки, полимер очищают трехкратным переосаждением гексаном из хлороформа. После чего, выделенные полимеры сушат в вакуумном шкафу при 40 С до постоянного веса.
6.2 Работа 2. Качественный анализ сополимеров ПБМА-ПФГ методом ИК-спектроскопии
Теоретическая часть
Как было рассмотрено выше, метод ИК-спектроскопии является не только качественным, но и количественным. Так, с помощью него можно определить состав сополимера, предварительно построив калибровочные зависимости соответствующих механических смесей различного соотношения. Например, ранее были определены составы сополимеров ПММА-ПФГ, ПNВП-ПФГ и других блок-сополимеров линейно-дендритного строения с использованием калибровочной кривой, построенной по полосам поглощения группC6F5 и полосам поглощения характерных для одной из групп виниловых полимеров.
Для построения калибровочной кривой на ИК-спектрах поглощения необходимо выделить полосы характерные для каждого из полимеров входящих в состав блок-сополимера. Так, в случае линейно-дендритного блок-сополимера ПNВП-ПФГ состав был определен по полосам поглощения групп — C6F5 (960 см-1, 1075 см-1, 1220 см-1) и полосам поглощения ПNВП. линейного и разветвленного полимеров в спектрах поглощения механических смесей ПNВП-ПФГ.
Экспериментальная часть
ЦЕЛЬ РАБОТЫ: Качественный анализ сополимеров ПБМА-ПФГ методом ИК-спектроскопии.
Аппаратура и оборудование
ИК-спектрометр Infralum-FT801, пленки полимеров. Перед измерением необходимо приготовить пленки методом полива из 8−10% растворов (в зависимости от ММ) в хлороформе. В ходе работы необходимо будет снять ИК-спектры ПБМА, ПФГ, выделенных блок-сополимеров ПБМА-ПФГ и проанализировать полученные спектры (соотнести полосы поглощения соответствующим группам линейного и разветвленного полимеров).
" Infralum-FT801″ .
Выполнение работы
- 1. Подключить прибор к персональному компьютеру.
- 2. Включить прибор кнопкой на левой боковой панели.
- 3. Прогреть прибор 10−20 минут (пока не замигает зеленая лампочка).
- 4. Перед началом работы создать папку, для сохранения ИК-спектров.
- 5. Снять опорный спектр (воздух).
- 6. В ячейке закрепить пленку полимера, вставить ее в прибор.
- 7. Снять спектр полимерной пленки ПБМА в режиме пропускания (выбрав режим Т-пропускание, после снятия спектра «убрать пики CO2», «нормировка 100%»).
- 8. Аналогичным образом снять пленки ПФГ, и блок-сополимеров.
- 9. Сравнить полученные ИК-спектры с библиотекой данных (выбрать
библиотеку данных и запустить поиск по спектру).
В отчете необходимо предоставить проанализированные ИК-спектры, с соотнесением характеристических частот группам сополимера.
6.3 Работа 3. Определение ММР сополимеров методом ГПХ. Анализ интегральных и дифференциальных кривых распределения
Теоретическая часть
При работе в методе ГПХ обычно используют рефрактометрический и спектрофотометрический детекторы. Принцип работы рефрактометрического детектора основан на измерении разности показателей преломления ?n элюата (раствора полимера) и элюента (растворителя), потоки которых после колонок попадают в сравнительную и рабочую камеры кюветы. Рабочая и сравнительная камеры представляют собой комбинацию двух смежных призм, разделенных прозрачной перегородкой и образующих плоскопараллельную пластину. Если через камеры проходят потоки жидкости с одним и тем же показателем преломления, например чистый тетрагидрофуран, то луч света, проходящий через камеры, не преломляется на границе их раздела. При этом регистрируется «нулевая» линия.
Если же через сравнительную камеру проходит элюент, а через рабочую — раствор полимера, у которого показатель преломления выше, т. е. существует разница показателей преломления ?n, то луч света на границе раздела камер преломляется и выходит под некоторым углом по отношению к своему начальному направлению, пропорциональным ?n. Рефрактометрический детектор является наиболее универсальным и широко используется, поскольку для полимеров практически всегда можно подобрать растворитель, в котором инкремент показателя преломления dn/dc будет достаточно велик (dn/dc? 0.1).
Для получения дополнительных данных о примесях полимера наряду с рефрактометрическим детектором может быть использован спектрофотометрический (фиксирует поглощение УФ-излучения в области = 254 нм, 340 нм — по двум каналам), который в систему подключается последовательно после рефрактометрического и регистрация данных может происходить одновременно с двух детекторов. Данный детектор является качественным, и никакой количественной информации не несет.
Также в последнее время широкое применение для анализа полимеров получили вискозиметрический детектор и проточный лазерный нефелометр (детектор малоуглового лазерного светорассеяния). Эти детекторы в комбинации с рефрактометром позволяет определять ММ сверхразветвленных полимеров, а также их степень разветвленности.
Определение ММР и средних ММ полимеров. Первичная информация о ММР получается в виде хроматограммы, которая представляет собой зависимость регистрируемого сигнала детектора от параметров удерживания — удерживаемого объема (Vr) или времени удерживания (tr). Хроматограмма — суперпозиция большого числа перекрывающихся пиков отдельных полимергомологов, высота сигнала на хроматограмме прямопропорциональна концентрации хроматографируемых макромолекул на выходе из колонки. Рефрактометрический детектор определяет массовую концентрацию полимера, поэтому хроматограмма дает массовую функцию распределения. Из которой с использованием калибровочной зависимости и соответствующих расчетов с помощью программного обеспечения LCSoluthon определяют значения средних молекулярных характеристик и ММР полимера в дифференциальной или интегральной форме.
Экспериментальная часть ЦЕЛЬ РАБОТЫ: Определить молекулярную массу сополимеров ПБМА-ПФГ методом ГПХ.
Аппаратура и оборудование
- 1. Хроматограф жидкостной LC-20 (Shimadzu).
- 2. Компьютер с программным обеспечением LСsolution (программное обеспечение LCSolution — GPC Software для обработки данных ММР).
- 3. Виалы с завинчивающимися крышками вместимостью 7−10 см3.
- 4. Весы аналитические.
- 5. Диски для фильтрации проб, диаметр 13 мм, с мембранами из политетрафторэтилена, размер пор 0.45 мкм.
- 6. Хроматографической шприц для ручного инжектора вместимостью 100 мм³ (объем шприца должен в 5 раз превышать объема петли в ручном инжекторе, который составляет 20 мм3).
Общее описания хроматографа LC-20
Хроматограф жидкостной модели LC-20 производства фирмы Shimadzu, состоит из насоса, инжектора для ручного ввода проб, рефрактометрического детектора RID-10A, спектрофотометрического детектора SPD-20AV, системного контроллера CBM-20A, термостата колонок CTO-20AC. Система сбора и обработки данных для жидкостного хроматографа: компьютер с программным обеспечением LСSolution (программное обеспечение LCSolution — GPC Software для обработки данных ММР). Комплект колонок для ГПХ серии GPC-800, например, GPC-801, GPC-8025, GPC-805.
Термостат.
Последовательность работы на хроматографе
- 1. Условия измерений. Измерения следует проводить при температуре воздуха от 15 до 30 С, атмосферном давлении от 84 до 106,7 кПа (от 630 до 800 мм рт. ст.), относительной влажности воздуха от 30 до 80%. Температура термостата колонок и ячейки рефрактометрического детектора 40 °C, объем вводимой пробы 20 мм³, скорость потока подвижной фазы 1 см3/мин, продолжительность регистрации хроматограммы 30 мин, режим работы рефрактометрического детектора — аналитический. Навеску полимера массой 0.010 гр. помещают в виалу с завинчивающей крышкой, добавляют 5 см³ ТГФ и оставляют до полного растворения полимера. Полученный раствор отфильтровывают через мембранный фильтр и помещают фильтрат в другую виалу. полимер синтез высокомолекулярный дифильный
- 2. Подготовка хроматографа и колонок, снятие хроматограмм. Каждый раз перед выполнением измерений проводят промывку всей системы и балансировку ячейки сравнения рефрактометрического детектора. Для этого последовательно включают все блоки хроматографа кнопками «power». Сначала загорается красная лампочка «error», далее, как только загорится зеленая «control» хроматограф готов к работе. Затем, открывают промывочный кран на 180 и промывают систему элюентом (ТГФ) в режиме «purge» кнопокой на блоке «Rid-10A», в ходе промывки на дисплее появляется «purging line» (скорость потока 1 см3/мин). По окончанию промывки закрывают промывочный кран. Далее открывают программное обеспечение LCSolution на экране появляется окно «Real Time Analysis», в меню «File» открывают «Open method file.» и выбирают метод по которому будут снимать хроматограмму (хроматограф откалиброван по полистиролу и по полиметилметакрилату), например, «PolySt+UV+kalib.lcm», далее нажимают «OK». После чего, кнопкой «Rid (Detector)R.Flow.on/off» промывают ячейку дифференциального рефрактометра в течение 2−3 минут. Далее нажатием клавиши «balance», дожидаются стабилизации значений на блоке Rid-10A. По окончании балансировки кнопка отключается автоматически и на блоке появляется «over» .
Обнуляем значения детекторов, А (спектрофотометр) и В (дифференциальный рефрактометр) кнопками «ZeroDetector A», «ZeroDetector B». Первой пробой всегда вводят чистый растворитель — ТГФ. Для закалывания пробы в петлю нажимают «Single start», появляется окно «Single run», где необходимо выбрать папку для сохранения хроматограммы, и записать название файла, после чего, открывают файл. Далее растворитель ТГФ с помощью шприца вводят в ручной инжектор, который поворачивается перед введением пробы до упора вверх, далее вкалывается проба и инжектор поворачивается до упора вниз, шприц остается в хроматографе.
В это время в окне «LC Running» ведется автоматическая запись хроматограммы. Время анализа составляет 30 минут, пик выхода растворителя соответствует 22.5 минутам.
Далее приготовленный и отфильтрованный раствор полимера анализируют аналогичным образом, только исключая промывку системы и ячеек.
3. Обработка хроматограмм. При анализе полимеров методом ГПХ время удерживания полимера зависит от его молекулярной массы. Первыми выходят пики, соответствующие соединениям с большей молекулярной массой, позже — соединения с меньшей молекулярной массой, и в конце анализа выходит растворитель.
Для обработки хроматограммы необходимо открыть программу заново в режиме «PostRun». Расчеты проводятся автоматически с помощью программного обеспечения GPC Software. Результаты определения молекулярно-массового распределения представляют в виде интегральной и дифференциальной кривой распределения.
6.4 Работа 4. Определение прочностных характеристик пленок сополимеров ПБМА-ПФГ
Порядок работы на универсальной испытательной машине «ZwikZ005» и ее устройство подробно описаны в учебно-методическом пособии «Механические свойства полимеров» (Составители: Смирнова Л. А. Мочалова А.Е., Учебно-методическое пособие. — Нижний Новгород: Нижегородский госуниверситет, 2011. — 21 с.).
Для изучения прочностных характеристик полимерных пленок ПБМА-ПФГ готовят 30% растворы полимеров в хлороформе. После чего, методом полива заливают пленки, и вырезают из них подложки пригодные для механических испытаний.
Полученные данные результаты представляют в виде диаграмм напряжение-деформация (например, рис. 7).
6.5 Работа 5. Получение изотерм поверхностного давления сжатие-расширение для дифильных полимеров
Теоретическая часть
Установка для изучения монослоев Ленгмюра и получения пленок Ленгмюра-Блоджетт «KSVmini» состоит из следующих основных блоков.
- — емкость, в которой находится жидкость (субфаза), называемая ванной;
- — поверхностные барьеры, движущиеся навстречу с постоянной скоростью по краям ванны;
- — электронные весы Вильгельми, для измерения величины поверхностного давления в монослое;
- — устройство перемещения подложки (Диппер);
- — блок управления.
Сама ванна обычно изготавливается из политетрафторэтилена (фторопласта), что обеспечивает химическую инертность и предотвращает возможность утечки субфазы. Материалом для изготовления барьеров служит полиацеталь. Термостабилизация осуществляется циркуляцией воды по системе каналов находящихся под дном ванны с помощью термостата.
Установка располагается на виброзащитном основании в специализированном помещении с искусственным климатом — «чистая комната». Все используемые химические реактивы должны быть предварительно осушены и перегнаны. Для измерения поверхностного давления в монослое в современных установках Ленгмюра-Блоджетт используется датчик поверхностного давления — электронные весы Вильгельми. Действие датчика основано на принципе измерения усилия необходимого для компенсации воздействия на пластинку Вильгельми силы поверхностного давления в монослое на границе раздела «субфаза-газ» .
Силы, действующие на пластинку Вильгельми, зависят от адгезии, парциального давления газа, плотности жидкости и др. (рис. 9).
W, l, t — ширина, длинна и толщина пластинки Вильгельми соответственно, h — глубина погружения в воду. Результирующая сила, действующая на пластинку Вильгельми, состоит из трех составляющих:
Материал пластинки Вильгельми выбирается таким образом, чтобы и = 0. Поверхностное давление это разность между силой действующей на пластинку, погруженную в чистую воду и силой действующей на пластинку погруженную в воду, поверхность которой покрыта монослоем:
Факторы, влияющие на качество пленок Ленгмюра-Блоджетт На качество пленок ЛБ влияют конструкционно-технологические факторы
(оборудование и технологическая чистота) и физико-химические факторы. Конструкционно-технологические факторы определяются совершенством современного оборудования, которое позволяет контролировать поверхностное натяжения в монослое, уровень субфазы, коэффициент переноса, характеризующий наличие дефектов в пленке ЛБ, температуру и pH-субфазы. Аналогичные требования предъявляются и к устройствам, которые удерживают подложку и перемещают подвижные барьеры (отсутствие механических вибраций, постоянство скорости перемещения образца, постоянство скорости перемещения барьера).
Все перечисленные факторы обеспечиваются:
— контролем чистоты исходных материалов (использование деионизованной воды со значением электропроводности не более 0.1 мкСм/см в качестве основы субфазы, приготовление растворов полимеров из реагентов квалификации ч.д.а. и непосредственно перед их применением);
- — проведением подготовительных операций, таких, как промывка подложек и барьеров по указанной методике;
- — предварительной очисткой поверхности субфазы;
- — обеспыливанием рабочей зоне и создание искусственного климата «чистая комната» (защитный бокс).
Физико-химические факторы определяются природой полимера, фазовым состоянием монослоя на поверхности субфазы, взаимосвязью между природой подложки, микрошероховатостью ее поверхности и качеством формируемой мультиструктуры, а также структурой молекулы, определяющей соотношение гидрофильных и гидрофобных взаимодействий между молекулами самого амфифильного полимера и молекулами субфазы, растворимость полимера в воде, химическими свойствами полимера (например, способность к образованию межмолекулярных ковалентных связей при внешнем энергетическом воздействии, приводящем к полимеризации, которая изменяет его механические и физические свойства), способность к образованию связей с металлами.
ЦЕЛЬ РАБОТЫ: Получение изотерм поверхностного давления в условиях сжатия-расширения дифильного полимера
Экспериментальная часть Приготовление растворов. Для получения изотерм поверхностного давления необходимо приготовить свежие растворы полимеров в хлороформе с рабочей концентрацией 1 мг/мл. Растворы готовят в пикнометрах на 3 мл. Далее рассчитывают объем посадки раствора полимера необходимый для снятия изотермы поверхностного давления исходя из ММ полимера, размеров молекул, площади ванны, с учетом того, что образование плотного мономолекулярного слоя происходит на площади половины ванны. В качестве субфазы используется деионизованная вода.
Перед работой необходимо на деионизаторе «Хроматек» получить деионизованную воду. Для этого необходимо:
- 1. Открыть кран подачи воды, опустив шланг в емкость для предгона;
- 2. Нажать кнопку «Вкл.» на передней панели прибора. Дождаться показания значения электропроводности менее 0.1 мкОм/см на дисплее передней панели прибора, после чего собрать деионизованную воду (на измерение одного раствора полимера (4−5 посадок) потребуется около 4 литров воды).
- 3. Выключение деионизатора: отключить подачу воды, после чего прибор отключиться автоматически. По завершению работы выключить прибор кнопкой «Выкл.» на передней панели прибора.
Подготовка установки для получения монослоев Ленгмюра с помощью пластинки Вильгельми
- 1. Перед использованием необходимо промыть тефлоновую ванну сначала деионизованной водой (400 мл), далее ацетоном (200 мл) и после полного высыхания — хлороформом (200 мл). После чего, опять промыть водой. Далее промыть подвижные барьеры, которые сделаны из полиацеталя, поэтому при их очистке используется только этиловый спирт (100 мл). При работе с тефлоновой ванной и барьерами необходимо использовать одноразовые перчатки. Все используемые в работе растворители должны быть предварительно осушены и перегнаны.
- 2. Далее устанавливают тефлоновую ванну и барьеры на подставку. После чего, заполняют ее деионизованной водой так, чтобы подвижные барьеры смачивались водой. Пластинку Вильгельми перед работой прокаливают в пламени горелки, и далее помещают на крючок в центре ванны. Ванну заполняют деионизированной водой. Включают термостат, для поддержания постоянной температуры субфазы (22 С).
- 3. Для определения степени чистоты ванны необходимо убедиться в том, что на поверхности воды отсутствуют примеси. Для этого сначала включают блок управления, далее открывают программное обеспечение
" KSVInstruments" (ярлык на рабочем столе «Sgserver»).
4. Открывают систему диагностики прибора («System Diagnostics and calibration»), при этом сигнал с установки для измерения монослоев через блок управления поступает на компьютер. После чего, закрывают это окно, и открывают ручное управление «Manual control unit». Обнуляют показания барьеров последовательным нажатием кнопок «balances», «zero», «barrier» ,.
" zero" в окне «Manual control unit». Кнопками на блоке управления сжимают барьеры, в результате чего, на дисплее в окне «Manual control unit» видны показания поверхностного давления. Далее барьеры останавливают кнопкой на блоке управления. Показания поверхностного давления должны быть 0.2 mN/m. Если поверхностное давление превышает данное значение, то заново промывают ванну водой, предварительно раздвинув барьеры (кнопкой на блоке управления). Для этого водоструйным насосом удаляют воду с поверхности ванны, предварительно обработав насадку на шланге спиртом, после чего опять наливают воду. После установления равновесия, обнулив показания, сдвигают барьеры и проверяют чистоту ванны. Если поверхностное давление после повторной промывки 0.2 mN/m, начинают измерения:
— микрошприцом наносят рассчитанный объем посадки раствора полимера в хлороформе на всю поверхность ванны по каплям (в разных точках ванны);
- — оставляют на 15 минут для испарения растворителя;
- — через 15 минут в левом верхнем углу открывают окно нажатием «fail», далее «New isotherm». Появляется окно управления ванной «Trough Controls», где необходимо задать следующие параметры: в методе сжатия («Compression Method») из выпадающего списка выбрать постоянную скорость сжатия («Constant rate compression»). В окне регистрации опций («Recording Options») выбирают «Manual», справа от которого нажимают «Rec on». В строке задания опций «Target Options» выбирают поверхностное давления «Surface Pressure», и устанавливают количество циклов — 1 (при снятии изотерм сжатия — расширения задают 2 цикла). Далее задают точные параметры сжатия («Сompression Parameters»), по которым необходимо снять изотерму сжатия: максимальное давление, до которого будет снята изотерма сжатия («Target», mN/m), обычно это значение около 30 мН/м), и скорость сжатия («rate», mm/min) = 10.
- — Перед запуском снятия изотермы необходимо обнулить все показания, для этого в окне управления ванной «Trough Controls» нажимают «ZeroBal», а в окне «Manual control unit» последовательно необходимо нажать «Balances», «Zero», «Barrier», «Zero». После чего запустить режим снятия изотерм кнопкой «Go» в окне «Trough Controls». На экране появиться изотерма в режиме «on-line». После снятия изотермы окно «Trough Controls» исчезает автоматически, посмотреть изотерму можно в главном меню в режиме просмотра. После окончания эксперимента
необходимо раздвинуть барьеры кнопкой на блоке управления, водоструйным насосом убрать воду из ванны, и промыть водой (возможно, эту процедуру придется повторить не один раз, поскольку все зависит от степени чистоты ванны, промывать ее необходимо до тех пор, пока показание поверхностного давления не будет 0.2 mN/m).
— Для получения изотермы сжатие — расширение на водную субфазу наносят тот же объем раствора полимера. Последовательность работы аналогичная, только в окне управления ванной необходимо задать значение поверхностного давления, после которого начнется расширение монослоя («Target», mN/m). Его определяют по изотерме сжатия, путем построения касательной к участку кривой, на котором наблюдается резкий рост поверхностного давления (т.е. это то давление, при котором монослой уже сформировался, но еще не разрушился). Также необходимо указать количество циклов = 2. После снятия изотермы сжатия-расширения необходимо также раздвинуть барьеры и промыть ванну.
В зависимости от природы полимера и объема наносимого на субфазу раствора вид изотерм может отличаться. Поэтому, как правило, эксперимент повторяют при различных объемах посадки раствора полимера. Из полученных изотерм Ленгмюра рассчитывают площадь, занимаемую одной молекулой полимера в плотном монослое.