Основы хроматографического метода анализа

Идентификацию разделяемых компонентов проводят преимущественно двумя методами: с использованием веществ-свидетелей и времени удерживания. Когда эти методы применить невозможно, для идентификации компонентов разделяемой смеси используют индекс удерживания Ковача 1. Он характеризует удерживание вещества X на колонке с определенной неподвижной фазой при температуре t°C относительно двух я-алканов… Читать ещё >
Основы хроматографического метода анализа (реферат, курсовая, диплом, контрольная)
Сорбция (от лат. sorbeo — «поглощаю») — поглощение твердым телом или жидкостью какого-либо вещества из ОС. Различают четыре вида сорбции.
Абсорбция — поглощение какого-либо вещества из ОС всей массой поглощающего тела (абсорбента). Адсорбция — поглощение вещества из газовой или жидкой среды поверхностным слоем твердого тела (адсорбента) или жидкостью. Хемосорбция — поглощение вещества твердыми или жидкими сорбентами с образованием химических соединений. Капиллярная конденсация — образование жидкой фазы в порах и капиллярах твердого сорбента при поглощении паров вещества.
По агрегатному состоянию фаз хроматографию разделяют на газовую, жидкостную и свсрхкритичсскую флюидную. Газовая хроматография включает в себя газожидкостную и газо-твердофазную, жидкостная — жидкостно-жидкостную, жидкостно-твердофазную и жидкостно-гелевую.
Газовая — хроматография, в которой подвижная фаза находится в состоянии газа или пара. Газожидкостная — газовая хроматография, в которой неподвижной фазой служит жидкость, нанесенная на твердый носитель. В ее основе лежит абсорбция газов и паров, т. е. поглощение жидкостью. В этом случае колонка заполнена шихтой — пористыми телами, которая является носителем неподвижной фазы (жидкости), абсорбирующей газы. Колонка представляет собой металлический капилляр с внутренним диаметром 0,3—0,5 мм и длиной 15—30 м, намотанный на катушку.
Жидкостная — хроматография, в которой подвижной фазой является жидкость. Ситовая (гельпроникающая или гельфильтрационная) — вариант жидкостной хроматографии, когда время выхода вещества из колонки зависит в основном от геометрических и диффузионных параметров разделяемых макромолекул. Высокоэффективная жидкостная хроматография (ВЭЖХ) — использование сорбентов с очень небольшим размером зерна (3—10 мкм), что обеспечивает быстрый массоперенос при очень высокой эффективности разделения.
Флюидная хроматография (сверхкритическая) — когда процесс протекает при сверхкритических условиях, вследствие чего газ-иоситель ведет себя подобно жидкости. В этом случае можно использовать колонки и детекторы, предназначенные для газовой хроматографии. Плотность же и растворяющая способность реагентов приближаются к соответствующим свойствам жидкости.
По механизму взаимодействия сорбента и сорбата выделяют несколько видов хроматографии: распределительную, основанную на различии в растворимости разделяемых веществ в неподвижной фазе (газожидкостная хроматография) или на различии в растворимости веществ в подвижной и неподвижной жидких фазах; ионообменную — на разной способности веществ к ионному обмену; сорбционную — на различии в адсорбируемости веществ твердым сорбентом; эксклюзионную — на различии в размерах и формах молекул разделяемых веществ; аффинную — на специфических взаимодействиях, характерных для некоторых биологических и биохимических процессов.
По технике выполнения выделяют колоночную и плоскостную хроматографию, когда разделение проводится на специальной бумаге (бумажная хроматография) или в тонком слое сорбента (тонкослойная хроматография).
По цели хроматографирования выделяют аналитическую, препаративную и промышленную (производственную) хроматографию. Аналитическая — хроматография, используемая для количественного и качественного анализа смесей. Препаративная — высокоэффективный, чаще всего лабораторный метод получения веществ в чистом виде, концентрирования и выделения компонентов или фракций из смеси. Аналитическая реакционная газовая хроматография — метод, в котором сочетают хроматографическое разделение и определение с химическим или физическим изменением состава смеси, причем все эти стадии единого процесса (включая детектирование) осуществляются в одном общем потоке газаносителя. Проявительная — хроматография, при которой дискретно вводимое ограниченное количество разделяемой смеси вымывается из хроматографической колонки потоком непрерывно проходящего газа-носителя, сорбирующегося слабее любого из компонентов смеси. Экстракционная хроматография — метод, в котором элементарным актом является экстракция компонентов смеси в органическую фазу, закрепленную тонким слоем на инертном носителе. Ионопарная (разновидность экстракционной) хроматография занимает промежуточное положение между ионообменной и адсорбционной (распределительной или обращепо-фазной) хроматографиями. Лигандно-обменная хроматография — метод, применяемый для разделения соединений, содержащих донорные гетероатомы или кратные связи. Он основан на образовании координационных связей между сорбентом и разделяемыми ионами или молекулами.
На рис. 5.1, а показана идеализированная хроматограмма смеси двух веществ. По оси абсцисс отложено время хроматографирования (можно отложить объем элюата), по оси ординат — аналитический сигнал, зависящий от концентрации веществ в элюате (отклик А).
ГОСТ 17 567–81 определяет следующие основные параметры хроматографического процесса.
Время удерживания tR — интервал времени от момента ввода пробы в хроматографическую колонку до момента выхода из нее определяемого вещества максимальной концентрации. Значение tR не зависит от количества пробы, но зависит от природы вещества и сорбента, а также плотности сорбента и может меняться от колонки к колонке, tR =tm +ts, где tm, ts — время пребывания вещества в подвижной и неподвижной фазах.
Расстояние удерживания lR — длина отрезка диаграммной ленты, соответствующая времени удерживания.
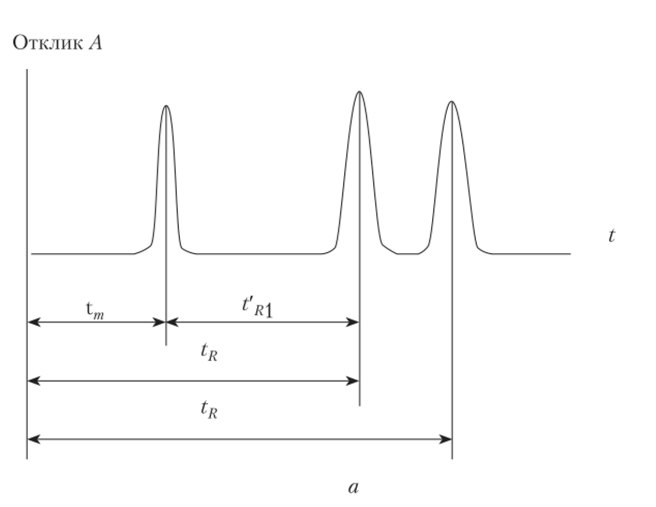
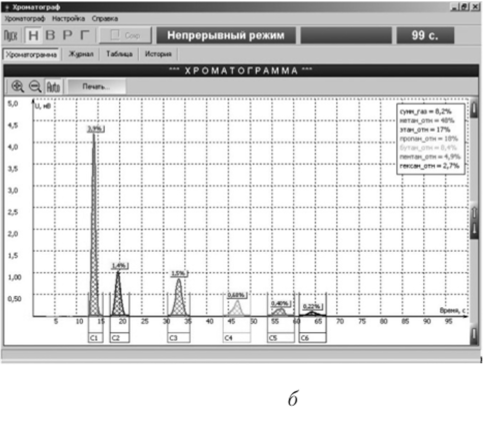
Рис. 5.1. Хроматограмма:
а — идеализированная хроматограмма двух веществ (пик с Ьщ соответствует неудерживаемому компоненту); б — окно компьютера с хроматограммой.

б.
Удерживаемый объем VR — объем газа-носителя, прошедшего через колонку от момента ввода пробы до момента выхода определяемого вещества максимальной концентрации, измеренный при давлении и температуре на выходе колонки,.

где Va — объемный расход газа-носителя при температуре и давлении на выходе колонки.
Время нахождения молекул исследуемого соединения в газовой фазе зависит от доли пустот в насадочной или капиллярной колонке. В разных насадочных колонках плотность набивки может изменяться, в результате будет меняться и величина времени удерживания tR. Поэтому для характеристики истинной удерживающей способности определяют величину приведенного времени удерживания.
Приведенное время удерживания t'R — интервал времени от момента выхода из колонки несорбирующегося вещества максимальной концентрации до момента выхода определяемого вещества максимальной концентрации,.

Относительное удерживание г — отношение приведенного времени удерживания определяемого компонента к приведенному времени удерживания вещества сравнения,.

где tRcp — время удерживания вещества сравнения; /ср — расстояние удерживания вещества сравнения; 1т — расстояние удерживания несорбирующегося вещества.
Параметры удерживания, по существу, характеризуют сорбционную способность анализируемых соединений. Отличие в сорбируемости определяется различием межмолекулярных взаимодействий «вещество — сорбент».
Любой процесс распределения вещества между двумя фазами характеризуется коэффициентом распределения D

где Ст и Cs — концентрация вещества в подвижной и неподвижной фазах соответствен но.
Для разделения двух веществ необходимо, чтобы D, * D2.
Коэффициент распределения D связан с хроматографическими параметрами следующим образом:
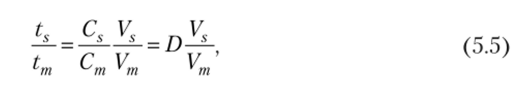
где Vs — объем подвижной фазы колонки. После ряда подстановок получим.

Исправленный объем удерживания VR связан с D соотношением.

Формулы (5.6) и (5.7) считают основными уравнениями хроматографии.
Эффективность хроматографической колонки — это расчетная величина, характеризующая степень расширения зоны определяемого вещества на выходе колонки, она пропорциональна квадрату отношения времени удерживания к ширине хроматографического пика. Количественно эффективность колонки можно оценить числом теоретических тарелок п и высотой, эквивалентной теоретической тарелке II. Между этими параметрами существует соотношение.

Для изотермической хроматографии эффективность определяют по числу теоретических тарелок п
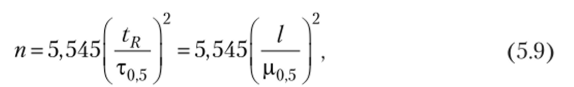
где т0 5 — ширина хроматографического пика, измеренная на половине его высоты и выраженная в единицах времени; р0 5 — ширина хроматографического пика, измеренная на половине его высоты и выраженная в единицах длины диаграммы регистратора.
Чем больше число теоретических тарелок, тем эффективнее работа хроматографической колонки. Их число может составлять от нескольких сотен до нескольких тысяч.
Предел обнаружения хроматографической колонки — наименьшее содержание контрольного вещества, определяемое хроматографическим методом с заданной доверительной вероятностью. Градуировочная хроматографическая характеристика — зависимость выходного сигнала от количества определяемого компонента, устанавливаемая опытным или расчетным путем и выраженная в виде формул, таблиц или графиков. Таким образом, род веществ определяют по времени удерживания в колонке, а концентрацию — по площади пика.
Идентификацию разделяемых компонентов проводят преимущественно двумя методами: с использованием веществ-свидетелей и времени удерживания. Когда эти методы применить невозможно, для идентификации компонентов разделяемой смеси используют индекс удерживания Ковача 1. Он характеризует удерживание вещества X на колонке с определенной неподвижной фазой при температуре t°C относительно двух я-алканов с числом углеродных атомов п и п + р. Индексы удерживания рассчитывают с помощью линейной интерполяции логарифмов исправленных параметров удерживания при соблюдении условия tR^ < tRm < tR(n + py
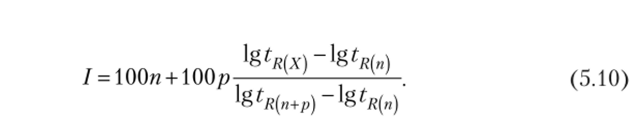
На практике применяют следующие методы расчета содержания компонентов: абсолютной градуировки (калибровки), внутренней нормализации и внутреннего стандарта. Все они основаны на измерении параметров пиков на хроматограмме: их площади или высоты. Чаще измеряют площадь пика (рис. 5.2).
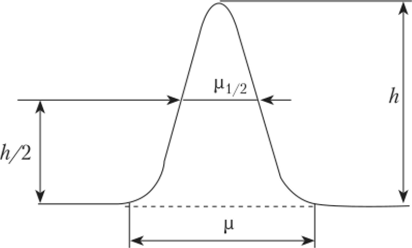
Рис. 5.2. Определение площади пика на хроматограмме
Количество вещества, вымываемого из колонки, можно найти по площади под кривой элюирования:

где С — концентрация вещества; V — объем.
Площади пиков на хроматограмме измеряют интегратором хроматографа. Это наиболее точный метод, так как ошибка измерения площади пика составляет менее 1%. При отсутствии интегратора площадь пиков S рассчитывают, измеряя их высоту и ширину или полуширину. В этом случае погрешности определения площади пиков достигают нескольких процентов: 
где р — ширина пика у его основания; р½ — полуширина пика; h — высота пика.
Основание пика на хроматограмме обычно несколько размыто, поэтому на практике измеряют не ширину пика р, а его полуширину pt/2— При таком способе рассчитанная площадь пика меньше его действительной площади на несколько процентов. К тому же пики на хроматограмме в той или иной мере несимметричны. Симметричность пиков, оцениваемая, например, на высоте, равной 0,1 высоты пика, может учитываться при расчете их площади.
При хроматографировании одновременно происходят разделение веществ и размывание хроматографических пиков разделяемых веществ, приводящее к ухудшению разделения.
Хроматографическое разделение основано на селективности сорбента и различии в термодинамических свойствах хроматографируемых веществ.
Разделение двух соседних пиков характеризуется разрешением Rs
и селективностью а
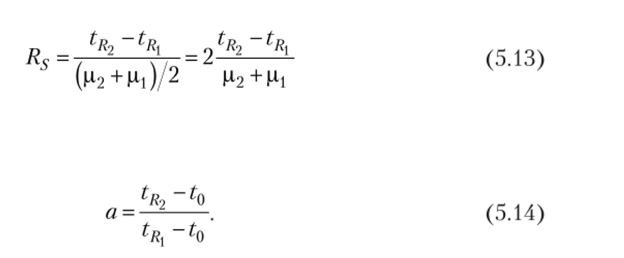
На рис. 5.3 показаны хроматограммы смеси двух веществ при разных эффективности колонки и селективности сорбента.
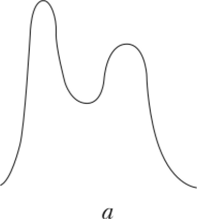
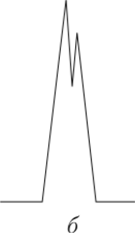
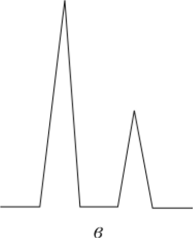
Рис. 53. Зависимость степени разделения смеси двух веществ от эффективности колонки и селективности сорбента:
а — высокая селективность, но плохая эффективность; б — высокая эффективность, но плохая селективность; в — высокая эффективность, достаточная селективность Температура очень сильно влияет на процессы хроматографического разделения. С ростом температуры увеличивается средняя скорость движения молекул в парогазовой фазе, в результате чего уменьшается разность скоростей между «убегающими» и «отстающими» частицами одного и того же компонента. Зоны разделяемых веществ (пик на хроматограммах) становятся более узкими, менее размытыми. В целом эффективность процесса разделения возрастает. Правда, с ростом температуры несколько снижается селективность хроматографической колонки.
При сравнительно низких температурах (200—250°С) разделяют и определяют относительно легколетучие вещества: некоторые углеводороды, спирты, эфирные масла. При более высоких температурах (250—400°С) разделяют и определяют фенолы, высокомолекулярные спирты, жирные кислоты.
Объем вводимой пробы зависит от специфики используемой методики и для жидких проб составляет 0,1 — 1 мкл. При большом объеме пробы обычно снижается эффективность хроматографической колонки.
Газохроматографические колонки представляют собой металлические или стеклянные трубки (прямые, изогнутые, спиральные) с внутренним диаметром 0,1—5 мм и длиной до нескольких метров (рис. 5.4). Они бывают наполненные (насадочные) и капиллярные.
Наполненные колонки — металлические (часто — из нержавеющей стали) или стеклянные трубки длиной 1—5 м, с внутренним диаметром 1,5—5 мм, обычно изогнутые в виде спирали. Эти трубки заполняются насадкой — частицами твердой основы с нанесением на их поверхность тонкого слоя неподвижной фазы.
Капиллярные колонки обычно представляют собой металлические или стеклянные (из кварцевого стекла) трубки. Неподвижная жидкость (вакуумная смазка) наносится на внутреннюю поверхность капилляра путем прокачивания ее раствора через капилляр и последующего испарения растворителя. Длина капиллярных колонок может составлять от нескольких десятков сантиметров до несколько сотен метров, внутренний диаметр — от 0,1 до 0,6 мм. Капиллярные колонки обеспечивают более высокую эффективность разделения многокомпонентных смесей.

Рис. 5.4. Хроматографические колонки:
а — наполненные; 6 — капиллярные В качестве поглотителей-сорбентов используют: оксиды кремния (силикагель, т. е. высушенная желатинообразная двуокись кремния) и алюминия, фторуглероды (тефлон, полихром), полистирол, цеолиты (алюмосиликаты щелочных и щелочно-земельных металлов), ионообменные смолы — иониты (органические полимеры, в макромолекулах которых содержатся ионизированные атомы или группы атомов, способные к ионному обмену), сополимеры стирола и дивинилбензола, а также стеклянные шарики, плавленый кварц, песок, графитированная сажа (карбохром), кристаллы хлорида натрия и т. д. Оптимальный размер зерен чаще всего колеблется в пределах 0,1—5 мм, удельная поверхность может составлять 100−1000 м2/г.
Неподвижная фаза представляет собой обычную нелетучую, высококипящую, с низкой вязкостью жидкость различной полярности и химической природы — углеводороды (гликоли или их смеси) с числом углеродных атомов в цепи от 10 до 30, полисилоксаны (силиконы), полигликоли (например, полиэтиленгликольадипипат), полиэфиры, амиды, амины, жирные кислоты и др.
Масса жидкой неподвижной фазы обычно составляет от 1 до 20% от массы твердого носителя (чаще всего — от 5 до 10%).