Примеры моделей в биоинформатике

Семейство матриц замещения РАМ. Исторически данные матрицы появились одними из первых, однако, несмотря на это, данное семейство матриц используются при анализе последовательностей до сих пор. Для построения матриц семейства РАМ используется эволюционный подход, учитывающий вероятность возникновения и последующего закрепления мутации (замены остатка). Согласно данной модели изменения в белках… Читать ещё >
Примеры моделей в биоинформатике (реферат, курсовая, диплом, контрольная)
Общие модели
Сравнение последовательностей биологических полимеров Точечные графики и матрица выравнивания
.Хотя процедура выравнивания для коротких последовательностей выглядит тривиально, в случае выравнивания последовательностей длиной в несколько сотен остатков, что соответствует типичной длине полипептидной цепи, или же тысяч остатков в случае последовательностей нуклеиновых кислот, а также при выравнивании последовательностей, содержащих редкие небольшие идентичные участки, задача существенно усложняется.
Первым способом сделать процесс выравнивания нагляднее было применение точечных графиков (dot-plots, рис. 3.1), где по осям отложены выравниваемые последовательности, а в местах пересечения строк и столбцов, сформированных идентичными остатками, располагаются точки.
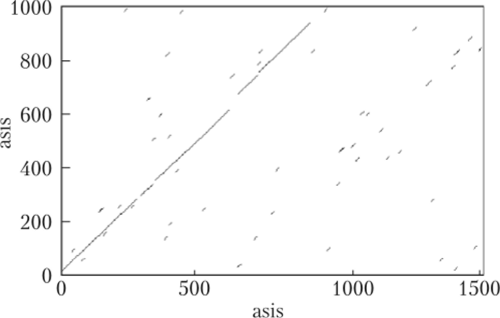
Рис. 3.1. Пример точечного графика для пары последовательностей
Таким образом, множество линий, параллельных главной диагонали, полностью соответствуют множеству локальных выравниваний соответствующей нары последовательностей. Множеству глобальных выравниваний на точечном графике соответствуют все способы провести линию из левого верхнего угла в правый нижний, используя участки, параллельные главной диагонали (выравнивание фрагментов из обоих белков), а также вертикальные (пропуск в горизонтально расположенной последовательности) и горизонтальные (пропуск в вертикально расположенной последовательности) фрагменты. Возможным локальным выравниваниям с пропусками на точечном графике соответствуют все возможные фрагменты глобальных выравниваний.
Точечный график позволяет наглядно отображать различные типы выравниваний, но при этом точечные графики обладают рядом недостатков:
- • невозможно проведение выравниваний в автоматическом режиме;
- • в случае выравнивания последовательности нуклеиновых кислот небольшой алфавит мономеров может приводить к высокой «зашумленности» точечного графика (рис. 3.2);
- • совьтравнивание разных типов аминокислотных остатков подразумевается одинаково «ценным», т. е. выравнивание двух аланинов и выравнивание двух триптофанов, что часто идет вразрез с биологическим смыслом, поскольку на самом деле совыравиивание более редких и специфичных аминокислот дает больше оснований предполагать наличие сходства между соответствующими позициями в белках;
- • график не позволяет учитывать выравнивание аминокислот, обладающих близкими физико-химическими свойствами, например гидрофобные и близкие по размерам валин, лейцин и изолейцин, хотя обычно подобного рода замены не приводят к дестабилизации структуры белка;
- • не производится дискриминация «ценности» выравнивания в зависимости от наличия, количества и длин пропусков в выравниваниях, хотя из общебиологических соображений очевидно, что наличие большого количества и (или) длинных пропусков свидетельствует о большем эволюционном расстоянии в случае парных выравниваний гомологичных последовательностей.
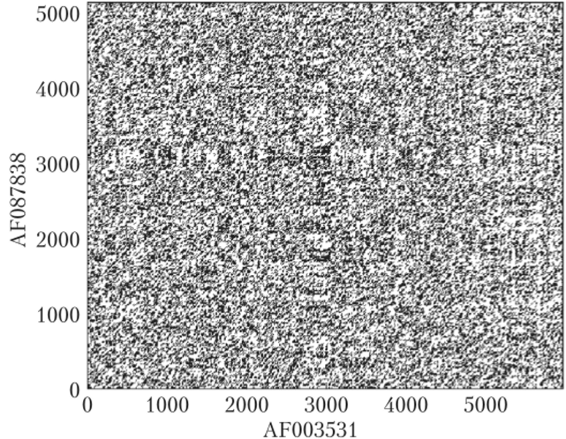
Рис. 3.2. Точечный график для последовательностей генов рецептора сульфонилмочевины Rattus norvegicus (AF087838) и Mus musculus (AF003531):
хотя нуклеотидные последовательности генов и имеют высокую гомологию, обнаружить ее на графике очень сложно Выравнивание на ориентированном графе. Решением проблем точечных графиков является их замена на граф выравнивания, введение матриц замещения и «штрафов» за пропуски в выравнивании. Как было упомянуто в гл. 2 (см. параграф 2.2), при выполнении выравнивания значения стоимости совмещения символов выравниваемых последовательностей берутся из матрицы замещения. Здесь мы несколько подробнее рассмотрим различные матрицы замещения, а также различные способы задания штрафов за пропуски в выравнивании.
Наиболее простой матрицей замещения является матрица идентичности (табл. 3.1), или унитарная матрица. В данной матрице стоимость замещения символа самого на себя равна единице, а все остальные позиции равны нулю. На самом деле в современном анализе белков подобная матрица практически не используется, разве что при построении точечных графиков. Однако следует отметить, что в задачах выравнивания нуклеотидных последовательностей унитарная матрица считается наилучшей.
Таблица 3.1
Унитарная матрица замещения.
А. | с. | D. | Е. | F. | G. | II. | I. | к. | L. | м. | N. | р | Q. | R. | S. | т. | V. | W. | У. | |
А. | ||||||||||||||||||||
С. | ||||||||||||||||||||
D. | ||||||||||||||||||||
Е. | ||||||||||||||||||||
F. | ||||||||||||||||||||
G. | ||||||||||||||||||||
Н. | ||||||||||||||||||||
I. | ||||||||||||||||||||
К. | ||||||||||||||||||||
L. | ||||||||||||||||||||
М. | ||||||||||||||||||||
N. | ||||||||||||||||||||
Р. | ||||||||||||||||||||
Q. | ||||||||||||||||||||
R. | ||||||||||||||||||||
S. | ||||||||||||||||||||
т. | ||||||||||||||||||||
V. | ||||||||||||||||||||
W. | ||||||||||||||||||||
У. |
Первой и, пожалуй, наиболее логичной с эволюционной точки зрения была матрица, построенная на схожести триплетов генетического кода.
(табл. 3.2). В этой матрице замещения различие между аминокислотными остатками выражалось в количестве изменений, которые необходимо внести в кодон одной аминокислоты, чтобы получить другую. Рассмотрим такие аминокислоты, как серин (кодон АГЦ) и цистеин (кодон УГЦ), поскольку для превращения одного кодона в другой нам достаточно сделать всего лишь одну замену в первом положении, то сходство между этими аминокислотами можно считать высоким. В то же время если мы возьмем аминокислоту аланин, которая часто кодируется триплетом ЦГУ, то для «превращения» ее в цистеин нам потребуется не менее трех модификаций, на основании чего можно заключить, что аланин и цистеин обладают достаточно низким сходством. Если в этой схеме учитывать частоты встречаемости различных кодонов, то можно получить достаточно точную матрицу замещения.
Таблица 3.2
Матрица замещения, основанная на генетическом коде[1]
А. | С. | D. | Е. | F. | G. | II. | I. | К. | L. | М. | N. | Р. | Q. | R. | S. | Т. | V. | W. | Y. | |
А. | ||||||||||||||||||||
С. | ||||||||||||||||||||
D. | ||||||||||||||||||||
Е. | ||||||||||||||||||||
F. | ||||||||||||||||||||
G. | ||||||||||||||||||||
Н. | ||||||||||||||||||||
I. | ||||||||||||||||||||
К. | ||||||||||||||||||||
L. | ||||||||||||||||||||
М. | ||||||||||||||||||||
N. | ||||||||||||||||||||
Р. | ||||||||||||||||||||
Q. | ||||||||||||||||||||
R. | ||||||||||||||||||||
S. | ||||||||||||||||||||
Т. | ||||||||||||||||||||
V. | ||||||||||||||||||||
W. | ||||||||||||||||||||
Y. |
В процессе развития методов выравнивания было создано огромное количество различных матриц замещения, однако наибольшее распространение получили семейства матриц РАМ и BLOSSUM.
Семейство матриц замещения РАМ. Исторически данные матрицы появились одними из первых, однако, несмотря на это, данное семейство матриц используются при анализе последовательностей до сих пор. Для построения матриц семейства РАМ используется эволюционный подход, учитывающий вероятность возникновения и последующего закрепления мутации (замены остатка). Согласно данной модели изменения в белках происходят за счет накопления некоррелирующих (независимых) мутаций, т. е. каждый остаток в последовательности белка эволюционирует независимо от других (рис. 3.3).
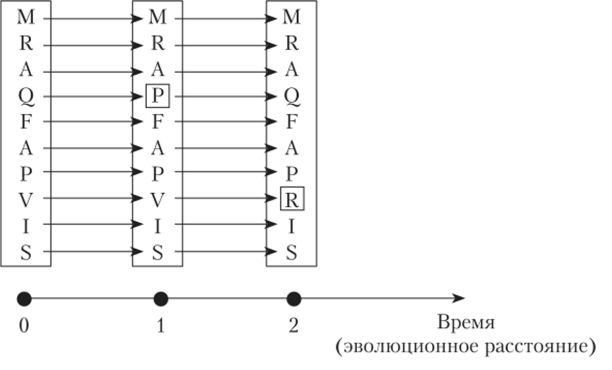
Рис. 3.3. Эволюционная модель Дейхофф, использовавшаяся при создании.
РЛМ-матриц замещения:
в ходе каждого элементарного эволюционного перехода каждый остаток может сохраниться либо измениться на другой с определенной вероятностью Термин РАМ в данном случае является сокращением от point (percent) accepted mutation, т. е. фиксированная точечная мутация — точечная мутация, закрепившаяся в процессе эволюции, и отражает степень эволюционного изменения, разделяющего две последовательности. Для получения вероятности замены одного остатка на другой использовались множественные локальные, без пропусков, выравнивания близких гомологов со степенью гомологии более 85% (рис. 3.4).
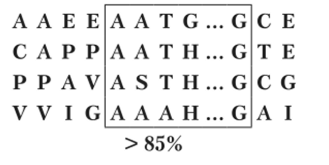
Рис. 3.4. Фрагмент множественного выравнивания последовательностей, использовавшийся для получения РАМ-матриц замещения:
высокая степень гомологии (> 85%) необходима для того, чтобы в каждой колонке было не более одной замены На основе выравниваний определялись предковые последовательности и строились эволюционные деревья. После подсчета количества замен в ветвях деревьев рассчитывались частоты замещений (рис. 3.5).
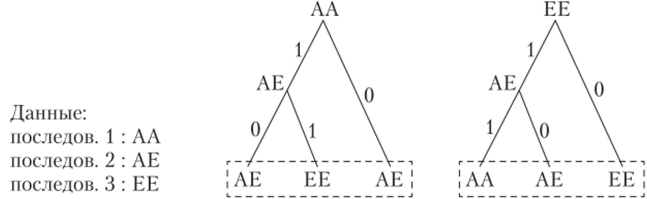
Рис. 35. Пример построения эволюционных деревьев для фрагментов трех последовательностей:
деревья строились таким образом, чтобы минимизировать количество замен в ветвях (:maximum parsimony tree)
Частоты замещений объединялись в матрицу переходов, которая показывает вероятность остатка не измениться или, наоборот, мутировать за определенный эволюционный интервал. После построения матрицы переходов все ее значения умножались на определенный коэффициент, подобранный таким образом, чтобы средняя вероятность мутации остатка с учетом данной матрицы была равна 1%. Итоговая матрица была названа матрицей переходов РАМ1 (рис. 3.6).
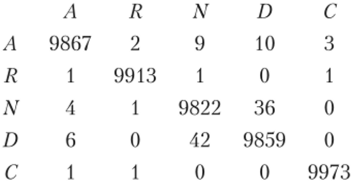
Рис. 3.6. Верхний левый угол матрицы переходов РАМ1:
для наглядности все значения умножены на 104 и округлены, г. е. вероятность аланина сохраниться в течение эволюционного интервала РАМ1 равна 0,9867, а вероятность его замены на цистеин равна 0,003.
Особенность матрицы переходов состоит в том, что для получения матриц переходов для больших эволюционных расстояний, т. е. РАМЛГ, в общем случае нам достаточно умножить матрицу переходов РАМ1 саму на себя N раз.
Для расчета матриц замещения на основе матриц переходов была использована формула (3.1):
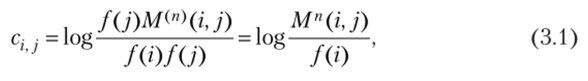
где i, j — номера остатков (от 1 до 20); с, у — элемент рассчитываемой матрицы замещения; Mn(i, j) — элемент матрицы переходов РАМн; /(/),/(/) — частоты встречаемости соответствующих остатков (г и j).
Данная формула представляет логарифм отношения двух выражений:
- 1) /0) ' М" (г, j) — вероятность встречи остатка j (J (/')), умноженная на вероятность того, что остаток j в данной колонке выравнивания или ветви эволюционного дерева получился из остатка i за эволюционное время РАМн (Mri(i, j)). Иными словами, это вероятность того, что произошедшая мутация является результатом нашей эволюционной модели;
- 2) /(/') • /(г) — произведение вероятностей случайной встречи остатков i и j в последовательностях белков. Произведение вероятностей в данном случае дает нам вероятность события комбинирования остатков i и j в одном столбце выравнивания в силу случайных причин.
Таким образом, логарифмируемое отношение показывает степень отличия вероятности перехода остатка i в остаток j по нашей модели от вероятности встретить такую ситуацию в выравнивании просто в результате случайности. Логарифм используется для того, чтобы у пас появилась возможность суммировать значения из матрицы замещения для оценки «неслучайности» выравнивания вместо взятия произведения, поскольку логарифм произведения равен сумме логарифмов множителей.
Семейство матриц замещения BLOSSUM. Данное семейство матриц было предложено в 1992 г., и в настоящее время матрицы этого типа применяются чаще всего. Для построения матриц семейства BLOSSUM используются множественные локальные выравнивания, однако способы анализа выравниваний разные. Название BLOSSUM является сокращением от BLOcks Substitution Matrices (блочные матрицы замещения). Алгоритм получения матриц не подразумевает эволюционного анализа, как в случае РАМ матриц, а использует частоты пар в так называемых блоках консервативных участков (рис. 3.7).
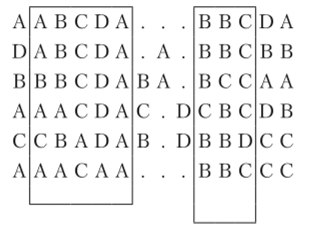
Рис. 3.7. Множественные выравнивания:
консервативные блоки выделены рамкой. Пример комбинирования пар для первого столбца первого блока: 6АА + 4АВ + 4АС + 1ВС + ОВВ + ОСС, всего 15 пар Каждая строка такого блока является участком одной из выравненных последовательностей, а каждый столбец — эквивалентной позицией для всех последовательностей. Далее путем комбинирования возможных пар остатков внутри столбца последовательно для всех столбцов и для всех блоков находились встречаемости для всех возможных пар остатков (/^) и, соответственно, определялись частоты этих пар {qtj)

где i, j — номера остатков (от 1 до 20). Далее рассчитывались частоты встречаемости индивидуальных остатков:

Приведем пример[2] расчета частот остатков для набора пар, представленного на рис. 3.7:
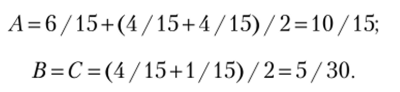
На основе полученных частот остатков, исходя из модели случайного комбинирования остатков в пары, рассчитывалась предполагаемая частота каждой пары равная произведению частот данных остатков при i = j и их удвоенному произведению при i * j. Далее вычислялся логарифм отношения наблюдаемых частот к предполагаемым исходя из случайной модели. Этот этап аналогичен расчету, но формуле (3.1) для РАМ-матриц:

Поскольку алгоритм получения матриц BLOSSUM не использует эволюционную модель, го для получения матриц, чувствительных к последовательностям с различной степенью идентичности, авторами было предложено использовать наборы блоков, характеризующиеся верхним порогом идентичности входящих в них последовательностей. Так, для построения матрицы BLOSSUM 62 — стандартной матрицы, предлагаемой в настройках «но определению» алгоритмом BLAST на сайте NCBI, — использовались блоки с коэффициентами идентичности составляющих их последовательностей не более 62%. Получение блоков с необходимым коэффициентом идентичности производилось путем кластеризации (объединения) нескольких последовательностей с высокими взаимными коэффициентами идентичности в единую последовательность с усреднением частот встречаемости остатков в них по колонкам блока. Иными словами, для упомянутой выше матрицы BLOSSUM 62 четыре верхние и самая нижняя последовательности левого блока на рис. 3.7 (ABCDA, ABCDA, BBCDA, AACDA и ААСАА) должны быть объединены в одну суперпоследовательность: (4/5А + 1/5В) (2/5А + 3/5В) С (1/5А + 4/5D) А. Они обладают взаимной гомологией более 62%, поскольку у данных последовательностей попарно идентичны четыре остатка из пяти — идентичность 80%.
Другой проблемой для создателей алгоритма была необходимость выбора начальной матрицы замещения, которая использовалась бы для получения «блоков» выравниваний. В данном случае проблема была решена применением матрицы идентичности в качестве исходной матрицы и построением матриц в несколько (три) итераций. Рассмотрим данную процедуру на примере получения матрицы BLOSSUM 80:
- 1) выбираем матрицу идентичности в качестве текущей;
- 2) получаем множественные локальные выравнивания с помощью текущей матрицы;
- 3) формируем блоки выравниваний с коэффициентом идентичности не более 80%, при необходимости производя кластеризацию последовательностей;
- 4) строим промежуточную матрицу замещения и выбираем ее в качестве текущей;
- 5) переходим к пункту 2 (повторить два раза) или к следующему пункту;
- 6) текущая матрица и будет матрицей BLOSSUM 80.