Описание модели квантово-химическими расчетами

Трудно переоценить роль молекулярной механики в современной химической практике. Поскольку все вычислительные проблемы относятся лишь к хорошо разработанным процедурам минимизации, даже на маломощных персональных компьютерах можно анализировать строение больших многоатомных молекул за разумное время. Возможность увидеть структуру молекулы на экране компьютера, рассмотреть ее с разных сторон… Читать ещё >
Описание модели квантово-химическими расчетами (реферат, курсовая, диплом, контрольная)
В разумности модели молекулы, используемой для квантово-химических построений, согласно которой анализу подлежит система ядер и электронов и ее поведение описывается уравнениями квантовой теории, сомнений нет. Вся совокупность экспериментальных данных, полученных разными методами, не противоречит этой модели. Трудности получения химически значимых результатов на ее основе связаны с тем, что она слишком общая и всеобъемлющая, так что численное решение уравнений представляет крайне сложную задачу. Приходится делать немалое число шагов на пути создания практичных алгоритмов расчетов свойств молекул, межмолекулярных комплексов и твердых тел.
Построение поверхностей потенциальной энергии (ППЭ) представляет важнейшую составную часть компьютерного эксперимента в химии, поскольку информация, содержащаяся в детальной картине этих поверхностей для молекулярной системы, поистине стоит тех серьезных затрат, которые неизбежны даже с применением мощной вычислительной техники.
Прежде всего на поверхностях потенциальной энергии находят стационарные точки, то есть координаты минимумов, максимумов, седловых точек. Для того чтобы можно было говорить о существовании стабильной молекулы или молекулярного комплекса, на потенциальной поверхности основного электронного состояния должен быть минимум, энергия которого меньше энергии любой совокупности фрагментов, на которые можно разбить молекулу. Если этих минимумов несколько, то для молекулы возможно несколько изомеров. Координаты ядер, отвечающие точкам минимумов, определяют равновесные геометрические конфигурации, а энергии по отношению к соответствующим пределам диссоциации на составные части — энергии связи химической системы. Знание положений и энергий седловых точек необходимо для оценок энергий активации при рассмотрении элементарных химических реакций. Наличие минимумов с энергией выше предела диссоциации указывает на возможность образования интермедиатов в системе реагирующих молекул. Рассчитывая разности электронных энергий различных электронных состояний для тех геометрических конфигураций ядер, которые отвечают точкам минимумов, можно интерпретировать или предсказывать электронные спектры молекул.
После аппроксимации фрагментов потенциальных поверхностей в окрестностях точек минимумов можно переходить к рассмотрению движений систем ядер молекулы и прогнозировать или интерпретировать колебательно-вращательные спектры. Зная набор электронных колебательно-вращательных энергий молекулы можно с помощью формул статистической термодинамики вычислять любые термодинамические функции данного вещества. Если рассматривается молекулярная система, в которой возможно перераспределение частиц, то есть химическая реакция, то рассчитывается сечение потенциальной поверхности вдоль пути наименьшей энергии, связывающего реагенты и продукты, и затем оценивается константа скорости элементарной химической реакции.
Описанная программа действий реализует схему расчетов свойств веществ без привлечения каких-либо эмпирических данных, которая представлена на рисунке 1.

Рисунок 1 — Реализация программы расчетов свойств веществ из первых принципов.
При этом результатом моделирования, которое немыслимо без компьютеров, является теоретический прогноз. Естественно, на любом промежуточном этапе этой схемы можно привлекать доступную экспериментальную информацию и вносить в компьютерное моделирование эмпирические элементы: при правильно сформулированной задаче ценность предсказаний не уменьшается, а становится более надежной. Расчеты геометрического строения и колебательных спектров молекул активно проводятся экспериментаторами, подтверждая результаты измерений квантово-химическим моделированием [4,5,6].
Молекулярная механика представляет собой совокупность методов априорного определения геометрического строения и энергии молекул на основе модели, в которой электроны системы явно не рассматриваются. Поверхность потенциальной энергии, которая в квантово-химических моделях подлежит прямому расчету, здесь аппроксимируется определенными эмпирическими функциями разной степени сложности, представляющими собой, например, суммы парных потенциалов взаимодействия атомов. Эти потенциальные функции, определяющие так называемое силовое поле молекулы, содержат некоторые параметры, численное значение которых выбирается оптимальным образом так, чтобы получить согласие рассчитанных и экспериментальных характеристик молекулы. В простейшем случае параметрами являются равновесные межъядерные расстояния и валентные углы, а также силовые постоянные, то есть коэффициенты жесткости упругих сил, связывающих пары атомов. Метод основан на допущении возможности переноса этих параметров из одной молекулы в другую, так что численные значения параметров, подобранные для некоторых простых молекул, используются далее при прогнозировании свойств других более сложных соединений.
Простейшие модели молекулярной механики учитывают растяжения связей, деформацию валентных и двугранных углов, взаимодействие валентно несвязанных атомов, называемое также Ван-дер-Ваальсовым взаимодействием, электростатические вклады и т. д.:
Для каждого слагаемого записывается определенное аналитическое выражение и параметры соответствующих функций подгоняются по каким-либо свойствам базовых молекул. Например, для описания потенциальной функции предельных углеводородов при не очень высоких требованиях к точности расчета достаточно около десяти параметров.
Сумма всех перечисленных вкладов определяет энергию U молекулы как функцию геометрической конфигурации ядер, и для нахождения равновесной геометрической конфигурации исследуемой молекулы необходимо определить минимум U с помощью компьютерной программы поиска стационарных точек на многомерных потенциальных поверхностях. Таким образом, практические действия исследователя чаще всего сводятся только лишь к заданию стартовой геометрии и вызову программы оптимизации геометрических параметров из условия минимума энергии. На выдаче просматривается полученная структура и. если необходимо, анализируются энергия и ее составляющие.
Трудно переоценить роль молекулярной механики в современной химической практике. Поскольку все вычислительные проблемы относятся лишь к хорошо разработанным процедурам минимизации, даже на маломощных персональных компьютерах можно анализировать строение больших многоатомных молекул за разумное время. Возможность увидеть структуру молекулы на экране компьютера, рассмотреть ее с разных сторон, проверить возникающие предположения о стерических затруднениях и т. д. оказывает неоценимую помощь в работе. Молекулярная механика играет роль молекулярного конструктора: для первичной оценки строения интересующей нас молекулы зачастую проще собрать молекулу на компьютере, чем тратить время на поиск необходимой информации в справочной литературе. При расчетах молекулярной структуры на более высоком уровне методами квантовой химии полезно использовать координаты ядер молекулы, найденные с помощью молекулярной механики, в качестве начального приближения. Для многих задач, например для конформационного анализа, уровень моделирования методами молекулярной механики оказывается вполне достаточным для качественных и даже количественных заключений.
В каждом конкретном случае необходимо интересоваться, для каких классов соединений параметризована та версия программы, которую предполагается применять при моделировании свойств нового соединения. Особенно осторожно следует относиться к оценкам энергий, хотя и для геометрических конфигураций возможны грубые ошибки. 5,6,7].
При моделировании методами молекулярной динамики или Монте-Карло интересующее нас свойство системы большого числа молекул вычисляется через статистические средние по положениям и движениям молекул. Как и в методах молекулярной механики, здесь также необходимо перечислить все частицы системы и задать потенциалы межчастичных взаимодействий. Однако в отличие от молекулярной механики в данных подходах области задания межчастичных потенциалов взаимодействия должны быть достаточно протяженными, и они не должны ограничиваться малыми смешениями от положений равновесия. Это накладывает существенно более высокие требования на способы расчета потенциалов.
Практически всегда уравнения, связывающие молекулярные параметры и свойства вещества, то есть макроскопические свойства, решаются численно, а эффективность решения существенно зависит от мощности используемых компьютеров. На рисунке 2 показаны схемы двух методик: Монте-Карло и молекулярной динамики, применяемых в компьютерных экспериментах. В обоих случаях задаются число молекул N, объем V, доступный для движения молекул, накладываются те или иные граничные условия, предписывается потенциал межмолекулярного взаимодействия U. В методе Монте-Карло обычно независимыми переменными, сохраняющими постоянные значения при моделировании, выбираются N, V и температура Т. Молекулы двигаются случайным образом в соответствии с предписаниями генератора случайных чисел, и каждое новое расположение либо принимается, либо отбрасывается с вероятностью, определенной по закону.
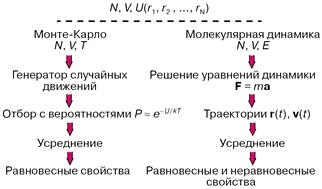
Рисунок 2 — Схема расчетов методами Монте-Карло и молекулярной динамики.
При моделировании в рамках молекулярной динамики положения r (t) и скорости v (t) каждой частицы в момент времени t определяются как решения системы уравнений классической механики (уравнений Ньютона) либо уравнений, в которых к силам F задаваемым потенциалом U, добавляются так называемые случайные силы. Макроскопические свойства рассчитываются при усреднении по положениям и скоростям молекул.
Как уже упоминалось, число частиц при моделировании методами Монте-Карло и молекулярной динамики с помошью современных суперкомпьютеров может достигать колоссальных величин. Даже без суперкомпьютеров достаточно типичны численные эксперименты для значений N порядка десятков и сотен тысяч. Примеры успешного применения методов Монте-Карло и молекулярной динамики для моделирования равновесных составов смесей при постоянном давлении, фазовых равновесий, адсорбции на поверхности твердых тел, свойств жидкостей в микропорах и т. д. достаточно многочисленны. Этими же методами решаются задачи поиска устойчивых конформаций (поворотных изомеров) полимерных молекул, чрезвычайно важные для биохимических приложений [5,6].
Рассмотрим достаточно последовательную квантовую модель на примере бимолекулярной реакции типа Х (i) + Y (j) > Х'(i') +Y'(j') + …
Здесь предполагается столкновение двух молекул X и Y, находящихся в состояниях i и j соответственно, которое приводит к продуктам реакции, то есть к молекулам X', Y',… в квантовых состояниях i', j', … Квантовая теория столкновений в принципе позволяет вычислить вероятности переходов между состояниями, отвечающими реагентам и продуктам, затем найти парциальные, то есть относящиеся к данным наборам квантовых чисел (здесь i, j, i', j',…), константы скорости. При усреднении по квантовым состояниям реагентов и продуктов можно оценить макроскопическую константу скорости соответствующей газофазной химической реакции как функцию температуры.
Полное осуществление этой программы в конкретных приложениях крайне затруднительно, даже если из предшествующих квантово-химических расчетов известна поверхность потенциальной энергии. Самой сложной стадией является численное решение уравнений квантовой теории столкновений с учетом перераспределения частиц, то есть как раз наиболее важная для химии стадия. Следует, однако, подчеркнуть исключительную важность научных исследований в этом направлении, поскольку они формируют каркас обшей теории, с которой сравниваются более простые модели. Кроме того, современные экспериментальные методы исследования динамики молекул позволяют измерить парциальные константы скорости и непосредственно сопоставить экспериментальные и теоретические результаты.
Более простые, а потому и более практичные способы вычисления констант скорости химических реакций получают обычно при определенных упрощениях полной квантовой модели. Так, начиная с 50-х годов проводятся компьютерные расчеты скоростей реакций методом классических траекторий. В этом методе, как и ранее, предполагается разделение электронной и ядерной подсистем, но в данном приложении необходимо знание поверхностей потенциальной энергии для достаточно широких интервалов межъядерных расстояний. Для расчета движений ядер, совместимых с данной потенциальной поверхностью, решают уравнения классической механики, а оценки констант скорости получают при сопоставлении числа траекторий, приводящих к реакции, с исходным числом траекторий при статистическом задании начальных условий.
На рисунке 3 изображены последовательные стадии вычислений методом классических траекторий. Реализация подобной схемы на современных компьютерах позволяет вычислять константы скорости реакций с существенно большим числом атомов, нежели при полностью квантовом описании [5,6].

Рисунок 3 — Схема расчетов констант скоростей химических реакций методом классических теорий.