Стадии поликонденсационных процессов

Приведенные значения констант скорости указанных реакций (при 30°С) свидетельствуют о существенной разнице в реакционной способности атомов Н в разных положениях фенольного кольца: к{ • 106 = 5,3; к • И)6 = 6,2; кг 106 = 8,7; кг 106 = 7,3; ?2″ Ю6 = 3,8; &з* 106 = 42,0; ку 106 = 9,1 лДмоль с). Соотношение между реакциями гомои гетерофункциональной конденсации при гидролизе кремнийорганичсских… Читать ещё >
Стадии поликонденсационных процессов (реферат, курсовая, диплом, контрольная)
Так же как и ценные, ступенчатые процессы синтеза макромолекул включают три основных стадии:
- • образование реакционных центров;
- • образование макромолекул (ступенчатый рост цепей);
- • прекращение роста цепей.
Образование реакционных центров
При использовании многих мономеров с ОН-, СООН-, КОСО-, С1СО-, Н2М-, НБи другими функциональными группами (см. табл. 4.1) реакционные центры уже имеются в составе указанных функциональных групп (например, атомы Н в ОН-, СООНи 1 ЬК-группах) и возможность их взаимодействия определяется лишь условиями — температурой, наличием катализатора и др.
Большинство поликонденсационных процессов являются каталитическими; катализатором в них является или один из исходных компонентов (например, дикарбоновая кислота), или специально вводимые вещества. По механизму действия катализаторов или инициаторов (механизму образования активных центров) В. В. Коршак предложил классифицировать поликонденсационные процессы на четыре основных типа:
- • катионная поликонденсация;
- • анионная поликонденсация;
- • ионно-координационные процессы;
- • свободнорадикальная поликонденсация.
Примерами поликонденсации, протекающей, но катионному механизму, могут быть реакции полиэтерификации и полиамидирования. Механизм реакции дикарбоновых кислот с гликолями, катализируемой протонами (их источником может быть как исходная дикарбоновая кислота, так и специально вводимая органическая или неорганическая кислота), включает протонировапие карбоксильной группы:
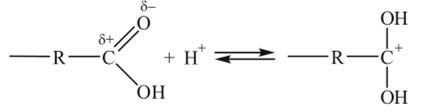
и последующую атаку ионом карбония ОН-группы гликоля: 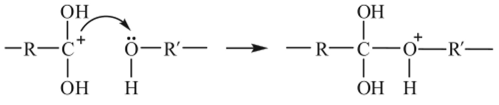
Оксониевый катион стабилизируется алюминированием протона, который, присоединяясь к одной из ОН-групп, способствует последующему отщеплению воды:
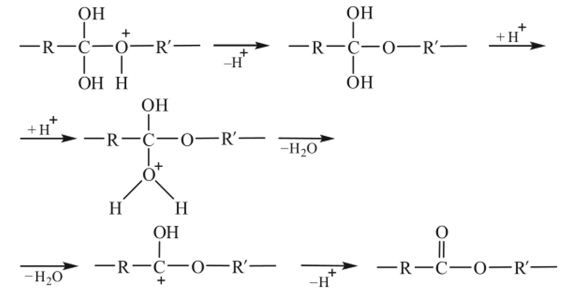
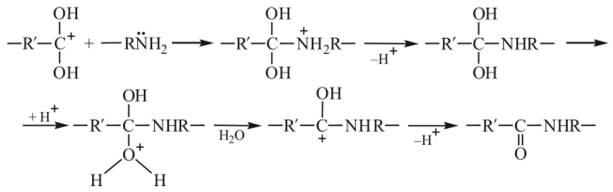
В случае полиамидирования процесс начинается с протонирования аминогруппы исходного мономера и взаимодействия образовавшегося аммонийного катиона с карбоксильной группой:
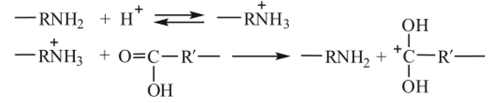
Дальнейшее превращение сформировавшегося реакционного центра — карбкатиона — аналогично приведенным выше для полиэтерификации:
Примером анионной поликонденсации является катализируемая гликолятами щелочных металлов полипереэтерификация эфиров дикарбоновых кислот гликолями: 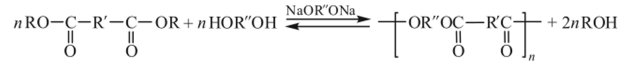
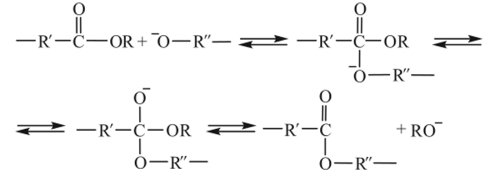
Образование активного центра в этом случае связано с атакой гликолевого аниона на атом углерода карбонильной группы и образованием оксониевого аниона, превращение которого с элиминированием аниона КО приводит к образованию межзвенной связи между реагирующими олигомерами:
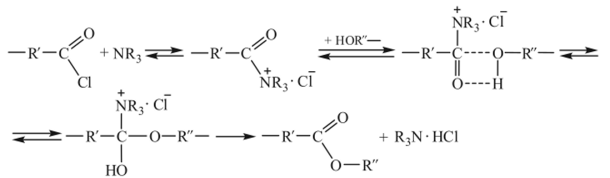
Ионно-координационный механизм характерен для процессов поликонденсации дихлорангидридов дикарбоновых кислот с бисфенолами в присутствии третичных аминов (синтез полиарилатов):
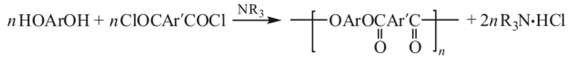
Формирование активных центров в этом случае связано с образованием ионной связи между молекулой третичного амина н хлорангидридной группой, последующей координацией соли с молекулой бисфенола и превращениями по схеме.
Во всех рассмотренных выше случаях функциональные группы в исходных мономерах присутствовали заранее и катализ процесса заключался лишь в переводе их в активное для данного взаимодействия состояние. Однако во многих случаях функциональные группы формируются непосредственно в поликонденсационном процессе.
Типичными примерами являются реакции образования радикалов в процессах полирекомбинации (см. схемы (4.14) и (4.16)): активные радикальные центры образуются непосредственно в процессе поликонденсации. Такая же ситуация характерна и для процессов образования фенолоформальдегидных полимеров (схема (4.10)), а также мочевино-, меламинои анилино-альдегидных олигомеров.
Поликонденсация фенолов с альдегидами активно протекает в присутствии ионных катализаторов — кислот, щелочей, солей органических оснований. Особенно активными являются щелочные катализаторы, в присутствии которых образуется феноксидный анион.
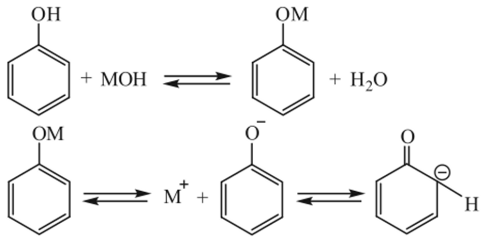
(М — атом щелочного металла), способный к перераспределению электронной плотности в ядре с локализацией ее в орто-положении, которое и атакуется молекулой альдегида ЯСОН:
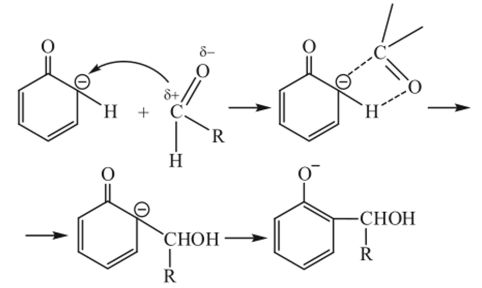
С близкой скоростью может идти замещение атома водорода и в пара-положении фенола: 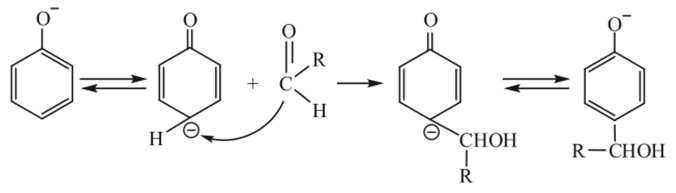
При избытке альдегида происходит дальнейшее замещение атомов водорода в цикле оили и-метилолфенолов. Схема возможных превращений при образовании триметилолфенола (в КСН=0 радикал И = Н) следующая:
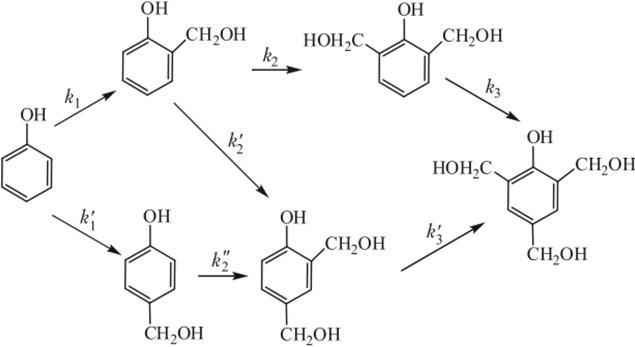
Приведенные значения констант скорости указанных реакций (при 30°С) свидетельствуют о существенной разнице в реакционной способности атомов Н в разных положениях фенольного кольца: к{ • 106 = 5,3; к • И)6 = 6,2; кг 106 = 8,7; кг 106 = 7,3; ?2" Ю6 = 3,8; &з* 106 = 42,0; ку 106 = 9,1 лДмоль с).
Электронодонорные метилольные группы, как правило, увеличивают способность фенола к дальнейшему замещению (кроме /7-метилолофенола); особенно активным является о, о-диметилолфенол (высокое значение к%).
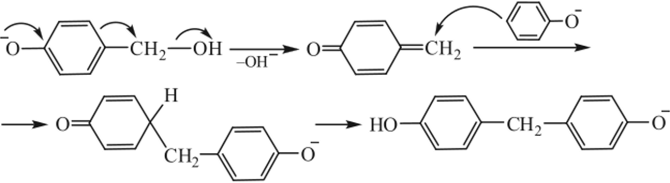
Неодинаковой реакционной способностью обладают и функциональные группы в формальдегиде (сам формальдегид и образовавшаяся из него метилольная группа). Как считают, взаимодействие с участием метилольных групп происходит следующим образом (на примере я-метилолфенола):
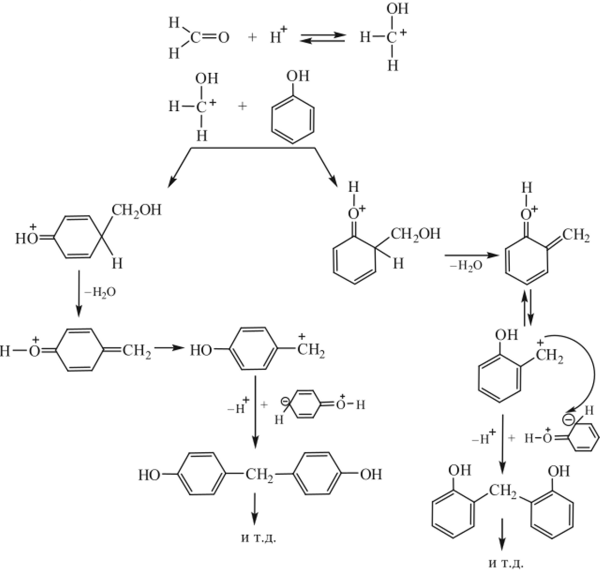
В присутствии кислотных катализаторов происходит протонирование альдегида и атака карбкатиона в ортоили пара-положение цикла по отношению к фенольному гидроксилу (дана схема для реакции фенола с формальдегидом).
Первый промежуточный продукт реакции — хинометидный катион — является очень активным и, быстро взаимодействуя с цвиттериоиной формой фенола, образует диоксидифенилметаи. В отличие от щелочного катализа в присутствии кислот метилольные производные фенола образуются редко, особенно при избытке фенола:
Характерным примером формирования активных центров непосредственно при поликонденсации является процесс гидролитической поликонденсации кремнийорганических мономеров типа Кш8{Х4_т, где Я — органический радикал (чаще СН3, С2Н5 и С6Н5); X — хлор, алкоксиили ацилолкси-радикалы; т = 1^-3. Для начала формирования цепи необходимо предварительное гидролитическое отщепление групп X с образованием связанных с атомом групп ОН, последующая конденсация которых и приводит к росту силоксановой цепи. В случае органохлорсиланов их гидролитическая поликонденсация может быть представлена схемой.
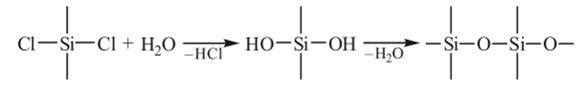
В определенных условиях (недостаток гидролизующего агента, основной катализ) кроме гомоконденсации силанольных групп становится возможной и гетерофункциональная поликонденсация:
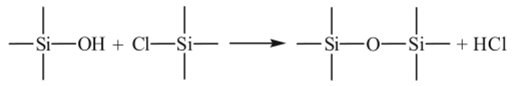
Соотношение между реакциями гомои гетерофункциональной конденсации при гидролизе кремнийорганичсских мономеров можно регулировать условиями процесса, а также изменением природы органического радикала и гидролизуемой группы в исходном мономере.