Иммунологические аспекты патогенеза апластической анемии

Flt3-лиганд (Flt3-L) — еще один цитокин, заслуживающий внимание при обсуждении механизмов развития АА. Flt3-L является фактором роста и дифференцировки для ранних гемопоэтических предшественников, дендритных клеток (ДК) и естественных киллеров (ЕК). Данные о нарушении лимфоцитопоэза и о дефиците зрелых ЕК у мышей с изменениями гена Flt-3-L доказывают роль данного цитокина в генерации популяций… Читать ещё >
Иммунологические аспекты патогенеза апластической анемии (реферат, курсовая, диплом, контрольная)
Иммунологические аспекты патогенеза апластической анемии
Апластическая анемия (АА) характеризуется панцитопенией, причиной которой является аплазия костного мозга (КМ). В КМ отмечается значительное уменьшение числа клеток-предшественников эритроидного, миелоидного и мегакариоцитарного рядов.
Количество CD34+ стволовых клеток составляет всего 1% от нормальных показателей. Кроме того, эти клетки функционально неполноценны — их колониеобразующая способность снижена по сравнению с клетками здоровых доноров [1]. Предшественники гемопоэза при апластической анемии также характеризуются крайне ограниченной способностью к усилению пролиферативной активности в ответ на воздействие рекомбинантных цитокинов (факторов роста) [3].
W. Zeng и соавт., изучая геном стволовых клеток больных апластической анемии, показали, что по сравнению со здоровыми донорами при апластической анемия вдвое увеличена экспрессия 805 генов, в том числе, генов апоптоза, подавления пролиферации, передачи сигналов, а также генов, кодирующих структуры цитокинов/хемокинов, участвующих в иммунном ответе (интерферон-g (ИФН-g), фактора некроза опухоли-a (ФНО-a), интерлейкинов (ИЛ) — ИЛ-6, ИЛ-8, ИЛ-1b, ИЛ-15, рецептора ИЛ-10, перфорин/гранзимов), и наоборот, значительно снижена экспрессия 238 генов факторов роста, транскрипции (GATA2, FLT3 рецептора), адгезии, а также факторов, участвующих в пролиферации и дифференцировке.
Это свидетельствуют о том, что оставшиеся немногочисленные CD34+ предшественники гемопоэза при апластической анемии не только лишены адекватной пролиферативной способности, но и подвержены апоптозу [4].
Кроме того, у части больных апластической анемии выявляются мутации гена, кодирующего теломеразу, что приводит к генетически обусловленным нарушениям механизмов сохранения теломер [5]. Ускоренная потеря и значительное укорочение теломеров хромосом стволовых клеток КМ и гранулоцитов, по сравнению с таковыми здоровых доноров аналогичного возраста, наблюдаемая при манифестации апластической анемии и стабилизирующаяся при достижении ремиссии заболевания, могут рассматриваться как доказательство акселерации апоптоза гемопоэтических клеток [1, 5, 6, 7, 8].
Иммунное подавление предшественников гемопоэза и их пролиферации иммунокомпетентными клетками (CD4+ и/или CD8+ Т-клетки, естественные киллеры — EK) и/или миелосупрессивными цитокинами представляет собой общепринятую теорию развития апластической анемии.
Однако роль CD4+ Т-хелперов и цитотоксических CD8+ Т-клеток в патогенезе апластической анемии недостаточно ясна. Также не известны антигены-мишени цитотоксических Т-эффекторов и поэтому, продолжается изучение Т-клонов и их специфичности.
Известно, что мононуклеарные клетки периферической крови больных апластической анемии подавляют рост и колониеобразующую способность не только аутологичных кроветворных клеток, но и аллогенных предшественников гемопоэза, полученных от пациентов с апластической анемии и от здоровых доноров. J. Maciejewski и соавт. и N. Zoumbus и соавт. высказали предположение о патогенетической роли CD8+ Т-клеток при данном заболевании [9, 10].
Иммунофенотипические исследования лимфоцитов КМ и периферической крови больных апластической анемии выявили снижение соотношения CD4+/CD8+ клеток у 73% больных апластической анемии за счет расширения CD8+ Т-клеточной популяции [10, 11] и повышенную экспрессию активационных антигенов CD25, CD38, CD71, HLA-DR [1, 11]. Однако тяжесть панцитопении не коррелирует с размерами CD8+ эффекторной популяции Т-клеток [12], что осложняет однозначную интерпретацию результатов.
По данным U. Moebius и соавт., клональная (клональность была доказана на основании изучения перестроек генов b-цепи Т-клеточных рецепторов (ТКР)) цитотоксическая популяция Т-клеток 8 из 18 больных апластической анемии имела CD4+CD8+ иммунофенотип и подавляла рост и дифференцировку предшественников гемопоэза и регрессировала после успешной иммуносупрессивной терапии. CD4+CD8+ фенотип характерен для Т-клеток на ранних этапах их созревания. У здоровых лиц CD4+CD8+ Т-клетки составляют 0,5−8% циркулирующих Т-лимфоцитов [13]. В отличие от этих результатов, S. Nakao и соавт. и W. Zeng и соавт. продемонстрировали хелперную природу (CD4+CD8- фенотип) Т-эффекторов при АА [14, 15]. Некоторые авторы сообщают, что исследование репертуара семейств гипервариабельных регионов b-цепи (Vb) ТКР при апластической анемии выявило олигоклональную природу иммунного ответа [16, 17]. Позже A. Risitano и соавт. уточнили, что для CD8+ Т-клеток характерна олигоклональность, а для CD4+ Т-клеток — поликлональность [18]. Однако это не позволяет отвергать важную роль Т-хелперов в развитии апластической анемии. Противоречивость этих данных можно объяснить крайней сложностью и разнообразием иммунных механизмов, участвующих в патогенезе каждого конкретного случая апластической анемии, а также тем, что апластической анемии является гетерогенной группой заболеваний, отличающихся по этиологическим и патогенетическим факторам.
Секвенирование гипервариабельных регионов (CDR3) b-цепей Т-клеточных рецепторов у больных апластической анемии в отличие от доноров выявило наличие специфических доминантных клонотипов (популяции Т-лимфоцитов с одинаковыми неслучайными изменениями CDR3), которые значительно регрессировали в результате иммуносупрессивной терапии. Более того, пациенты с идентичными гаплотипами молекул главного комплекса гистосовместимости (ГКГС) имели гомологичный доминантный CDR3 клонотип Vb семейств ТКР [19]. Поскольку CDR3 последовательность является «отпечатком» специфического патогена, можно считать, что наблюдения А. Risitano и соавт. подтверждают патогенетическую роль Т-олигоклонов при апластической анемии Наличие множественных Т-клеточных олигоклонов у больных апластической анемии можно объяснить тем, что иммунный процесс при апластической анемии спровоцирован разными антигенами. Кроме того, предшествующие гемотрансфузии или присутствующие инфекционные агенты могут вызывать возникновение олигоклонов. Так как в исследовании А. Risitano и соавт. изучали репертуар ТКР у больных без инфекционных осложнений и до начала гемотрансфузий, что исключает участие данных факторов в возникновении олигоклонов, можно предполагать роль множественных патогенов в развитии данного заболевания [19].
Изучение продукции цитокинов при апластической анемии позволило выяснить роль разных субпопуляций Т-клеток в развитии данного заболевания. У больных апластической анемии отмечается значительное повышение секреции миелосупрессивных цитокинов — ИФН-g и ФНО-a стромальными клетками КМ и периферическими мононуклеарными клетками, и что данные цитокины играют важную роль в патогенезе апластической анемии. Показано, что добавление анти-ИФН-g антител в культуру клеток КМ больных апластической анемии увеличивает образование колоний гемопоэтическими предшественниками [14, 20]. По данным E. Sloand и соавт. [21], у 51% нелеченных больных тяжелой апластической анемии отмечалась повышенная продукция ИФН-g циркулирующими Т-клетками. 96% больных с высокой экспрессией ИФН-g ответили на терапию, что сопровождалось снижением количества ИФН-g-содержащих Т-клеток, в то время как только у 32% пациентов с нормальной экспрессией ИФН-g лечение оказалось эффективным. Определение внутриклеточного ИФН-g, по мнению авторов, может служить прогностическим фактором [21].
Cтромальные клетки продуцируют ИФН-g [22]. Учитывая данные K. Kaito и соавт. о том, что экспрессия адгезивных молекул CD49d и CD49e на стволовых и стромальных клетках КМ значительно повышена у нелеченных больных апластической анемии, по сравнению со здоровыми донорами и с ответившими на лечение пациентами [23], можно предположить, что адгезивные молекулы способствуют осуществлению высокоаффинной связи между стволовыми клетками и эффекторами иммунной агрессии, осуществлению миелосупрессивного действия ИФН-g и развитию аплазии кроветворения.
Миелосупрессивный эффект ИФН-g сложен и осуществляется путем подавления клеточного цикла и индукции апоптоза гемопоэтических клеток. ИФН-g вызывает активацию множества внутриклеточных эффекторных молекул, влияющих на клеточный цикл, включая молекулы регуляторного фактора интерферона-1 (РФИ-1) [24]. Кроме того, повышение продукции двуокиси азота гемопоэтическими клетками [14, 25] и экспрессии Fas-молекул на CD34+ клетках КМ, а также активация р38 Map (митоген-актвированной протеин)-киназного сигнального каскада [20] интерфероном-g приводят к апоптозу предшественников гемопоэза при АА.
A. Verma и соавт. показали, что добавление ингибитора р38 Map-киназы в ко-культуры мононуклеарных клеток крови и предшественников гемопоэза больных апластической анемии в 100% случаев значительно увеличило формирование колоний. Авторы предполагают, что фармакологическое подавление активации р38 может служить эффективной терапевтической альтернативой для пациентов с апластической анемии [20].
Показано, что ИФН-g повышает уровень секреции ФНО-a макрофагами [20], а ФНО-a аутокринным механизмом увеличивает секрецию ИФН-g и чувствительность гемопоэтических клеток к эффектам ИФН-g [26]. Следует отметить, что данным цитокинам свойственен синергизм в супрессии кроветворения.
Flt3-лиганд (Flt3-L) — еще один цитокин, заслуживающий внимание при обсуждении механизмов развития АА. Flt3-L является фактором роста и дифференцировки для ранних гемопоэтических предшественников, дендритных клеток (ДК) и естественных киллеров (ЕК). Данные о нарушении лимфоцитопоэза и о дефиците зрелых ЕК у мышей с изменениями гена Flt-3-L доказывают роль данного цитокина в генерации популяций иммунокомпетентных клеток [27]. Показано, что уровень Flt-3-L в сыворотке больных апластической анемии в 30 раз превышает аналогичный показатель здоровых лиц [28]. Кроме того, достижение ремиссии при апластической анемии сопровождается снижением, а рецидив — повышением его уровня. Это свидетельствует о том, что Flt-3-L, с одной стороны, является частью компенсаторных механизмов, направленных на расширение кроветворения [28, 29]. С другой стороны, Flt-3-L способствует экспансии популяции зрелых ДК и ЕК, которые также продуцируют ИФН-g и следовательно, развитии аплазии кроветворения. Кроме того, воспалительные цитокины (ИФН-g, ФНО-б) продуцируются T-хелперами 1-го типа, которые являются ключевым звеном клеточного иммунного ответа, а также цитотоксическими Т-лимфоцитами и ЕК. T-хелперы 2-го типа секретируют ИЛ-4, ИЛ-5, ИЛ-9, ИЛ-10, ИЛ-13 и регулируют гуморальный иммунитет [30, 31, 32]. Продукция ИФН-g, ИЛ-10 и ИЛ-12 ДК, а также взаимоподавляющее действие Тh1 и Th2 хелперов осуществляют сложный баланс между данными популяциями Т-клеток.
Недавно опубликованное исследование по изучению апластической анемии показало, что in vivo Т-клеточный иммунный ответ 1-го типа ассоциируется с тяжестью заболевания [21], поэтому представляется целесообразным коротко рассмотреть пути развития Тh1 или Th2 иммунного ответа.
Известно, что Тh1-специфический фактор транскрипции — Т-bet экспрессируется Тh1-хелперами, CD8+ Т-клетками и естественными киллерами (ЕК). Т-bet повышает продукцию ИФН-g, а также увеличивает экспрессию b2-субединицы рецептора ИЛ-12. При этом, зависимость секреции ИФН-g от уровня транскрипции и экспрессии Т-bet более выражена у CD4+ Тh-клеток и у ЕК, чем у CD8+ T-клеток [30]. Кроме того, ИФН-g сам способствует транскрипции Т-bet гена [30, 31] посредством активации STAT1 (Signal Transducer and Activator of Transcription 1 — преобразователь сигнала и активатор транскрипции-1).
Продукция ИФН-g уже комитированными Тh1-клетками может быть индуцирована двумя путями — цитокин зависимым (ИЛ-12 и ИЛ-18) и путем передачи сигнала через Т-клеточные рецепторы. Исследования in vivo доказали, что комбинация ИЛ-18 и ИЛ-12 может увеличивать продукцию ИФН-g даже в отсутствии сигнала от ТКР [33]. При этом секреция ИФН-g, индуцированная цитокиновым путем, осуществляется посредством активации STAT4 и нуклеарного фактора-k В (НФ-kВ). Учитывая, что циклоспорин, А является ингибитором активации нуклеарного фактора активированных Т-лимфоцитов (НФАТ) и не влияет на цитокин-зависимый механизм продукции ИФН-g [30, 34], можно предполагать, что у той части больных АА, у которых терапия циклоспорином, А неэффективна, секреция данного миелосупрессивного цитокина регулируется ИЛ-12 и ИЛ-18 -зависимым путем.
В отличие от цитокин-зависимого пути, механизм передачи активационных сигналов через ТКР менее зависим от STAT4 и подразумевает активацию НФАТ. Следовательно, в тех случаях апластической анемии, в патогенезе которых участвует механизм активации НФАТ, терапия циклоспорином, А позволяет достигнуть положительных результатов [30, 34].
Следует также отметить, что активированные Т-лимфоциты продуцируют ИЛ-2 и экспрессируют рецепторы ИЛ-2. Данный цитокин не влияет на Тh1 или Th2 направленность иммунного ответа. ИЛ-2, действуя аутои паракринным путями, способствует росту и пролиферации всех субпопуляций Т-клеток. Однако, принимая во внимание более высокий порог активации Тh2 хелперов [31], а также наличие более благоприятных условий для экспансии Th1 хелперов (наличие ИФН-g, который способствует экспансии Th1 лимфоцитов и подавляют пролиферацию Th2 хелперов) при апластической анемии, можно предположить, что ИЛ-2 играет роль в преимущественной экспансии Тh1 хелперов и цитотоксических Т-клеток больных апластической анемии.
Таким образом, определение цитокинового профиля Т-хелперов и, следовательно, иммунного ответа представляет собой сложный процесс, от которого зависят особенности течения заболевания и эффективность лечения. Для апластической анемии характерно повышение соотношений Th1/Th2 популяций Т-клеток. Активация Th1-хелперов в ответ на презентацию экзогенных или эндогенных антигенов ДК или другими антиген-презентирующими клетками (АПК) может служить начальным этапом иммунного процесса при АА, приводящим к активации CD4+ и CD8+ клеток, к повышению продукции ИФН-g и ФНО-a и к деструкции гемопоэтических клеток.
N. Giannakoulas и соавт. исследовали цитокиновый профиль 24 больных апластической анемии до иммуносупрессивной терапии и в период длительной ремиссии. У нелеченных больных отмечалось преобладание CD4+ и CD8+ Т-клеток, продуцирующих ИФН-g и ИЛ-2 без стимуляции митогеном, и повышение ИФН-g/ИЛ-4 и ИФН-g/ИЛ-10 соотношений, что служит доказательством того, что при апластической анемии преобладает Тh1 тип иммунного ответа [35]. При длительной ремиссии количество CD8+ Т-клеток, продуцирующих ИФН-g, было снижено по сравнению с их уровнем, выявленном при манифестации заболевания, но, оставалось повышенным по сравнению со здоровыми лицами, причем, число ИФН-g секретирующих CD4+ Т-клеток также оставалось высоким [35]. При этом в период ремиссии процент Т-клеток, продуцирующих ИЛ-10, был значительно повышен, что обусловливало нормализацию соотношения Тh1 и Тh-2 клеток, секретирующих ИФН-g и ИЛ-4, соответственно, а также уменьшение соотношения популяции Тhh-1 хелперов, продуцирующих ИФН-g и CD4+ Т-клеток, секретирующих ИЛ-10 [35].
Таким образом, в период ремиссии у больных апластической анемии сохраняется персистенция Th1-хелперного иммунного ответа [14, 35, 36], что компенсируются повышенной продукцией иммуносупрессивных цитокинов — ИЛ-4 и ИЛ-10. Такой тонкий баланс легко может быть нарушен в случае нового антигенного стимула, приводящего к рецидиву заболевания.
Японские авторы описали два клинических случая апластической анемии, которые сопровождались повышением количества и активности естественных киллеров с фенотипомCD56+CD16 внутриклеточный+ИФНg+CD3-FasL- [37]. Длительное наблюдение в одном из случаев показало, что популяция ЕК уменьшилась при достижении ремиссии и увеличилась в момент развития рецидива апластической анемии. В результате данного исследования авторы высказали гипотезу о роли естественных киллеров в аплазии кроветворения. Однако этот вопрос требует подробного изучения.
Учитывая механизмы созревания и дифференцировки ЕК, а именно то, что в условиях непосредственного контакта стромальных клеток КМ с CD34+ стволовыми клетками Flt3-L и ИЛ-15 вызывают дифференцировку ЕК и приобретение характерных морфологических, фенотипических и функциональных (цитотоксичность, продукция цитокинов и хемокинов) качеств [27], а также что провоспалительные цитокины способствуют активации ЕК, можно предполагать, что повышенная продукция Flt3-L и ИЛ-15 стромальными клетками, а также секреция ИЛ-2, ИЛ-12, ИЛ-18, ИФН-g ДК и лимфоцитами при апластической анемии способствуют пролиферации, дифференцировке и активации ЕК [27, 30, 38]. Однако роль ЕК, которые могут распознать антиген еще до генерации антиген-специфических Т-клеток, в развитии апластической анемии остается неясной. Следует выяснить, имеет ли активация данной популяции специфический характер, или представляет собой вторичное явление, развившее на фоне всех вышеперечисленных изменений, наблюдаемых при апластической анемии.
Этиопатогенетические факторы, вызывающие возникновение цитотоксических Т-клонов и продукцию миелосупрессивных цитокинов, приводящих к развитию апластической анемии, неясны. Так как жизнеспособность, пролиферация и дифференцировка предшественников гемопоэза зависят от костномозгового микроокружения (стромальных клеток и продуцируемых ими ростовых факторов), аплазию кроветворения объясняли дефектом стромальных клеток КМ больных апластической анемии. Данная теория отвергнута как в ходе клинических наблюдений при эффективной трансплантации аллогенного КМ [1, 8], так и в экспериментальных исследованиях.
Показано, что in vitro стромальные клетки пациентов с апластической анемии поддерживают колониеобразование из CD34+ стволовых клеток здоровых лиц тогда, как формирование гемопоэтических колоний из CD34+ клеток больных апластической анемии в условиях нормальной стромы полностью подавлено [8]. In vitro культивирование стромальных клеток КМ больных апластической анемии и здоровых доноров в присутствии метилпреднизолона, снижающего уровень продукции миелосупрессивных цитокинов стромальными клетками, или без него показало, что в присутствии метилпреднизолона стромальные клетки КМ больных апластической анемии не отличаются от клеток контрольной группы по способности взаимодействовать с нормальными стволовыми клетками [39].
При изучении продукции гемопоэтических ростовых факторов стромальными клетками КМ показано, что уровни эритропоэтина, тромбопоэтина, гранулоцитарного колониестимулирующего фактора (Г-КСФ) (8) и Flt-3-лиганда [8, 27] в сыворотке крови при апластической анемии обычно повышены. Однако у небольшого количества больных апластической анемии наблюдается сниженная секреция гранулоцитарно-макрофагального колониестимулирующего фактора (ГМ-КСФ), Г-КСФ, ИЛ-3 [1]. Роль дефицита цитокинов в развитии данной болезни не выяснена.
Непосредственное повреждение клеток КМ в результате воздействия токсических веществ (метаболиты медикаментов, бензин) рассматривается как возможный механизм развития апластической анемии. Промежуточные продукты метаболизма ковалентно связываются с белками и ДНК клеток КМ. Генетическая вариабельность ферментов, ответственных за пути деградации таких веществ, может способствовать накоплению промежуточных метаболитов, которые сами по себе или после связывания с клеточными белками распознаются АПК и приводят к возникновению редкой идиосинкразии. Например, бензин метаболизируется в КМ, соединяется с белками и ДНК клеток КМ и вызывает их повреждение и апоптоз [8, 40]. Однако в настоящее время развитие апластической анемии редко ассоциируется с бензином.
Редки идиосинкразические реакции в виде аплазии кроветворения после применения лекарственных препаратов [8]. Сообщается о связи развития апластической анемии с приемом таких медикаментов, как тиазидные диуретики, сульфаниламиды, мебендазол и, в меньшей степени, противосудорожные и нестероидные противовоспалительные препараты [2, 8]. Контролируемые исследования выявили, что случаи развития апластической анемии связанные с приемом медикаментов в Таиланде, составляет всего 5%, что 5 раз меньше аналогичного показателя в Европе и Израиле [2].
Хорошо известна гепатит-ассоциированная апластической анемии (ГА-АА), развитие которой связывают с вирусами гепатитов, в первую очередь, вирусами гепатитов В и С. апластической анемии является редким осложнением вирусного гепатита, с риском развития 0,1−0,2% [41]. Доказанный острый вирусный гепатит предшествует развитию апластической анемии в 2−5% случаев в странах Европы и Северной Америки, а в странах Азии и Африки аналогичный показатель составляет 4−10%. Сообщалось об ассоциации вируса гепатита G с гепатит-ассоциированной апластической анемии [42]. Однако инфицирование вирусом гепатита G, скорее всего, связано с множественными гемотрансфузиями как при апластической анемии, так и при ГА-АА [41, 43].
Иммунопатологические механизмы, провоцирующие аберрантный иммунный ответ и приводящие к поражению КМ при ГА-АА, остаются мало изученными. ДК способны обеспечить антиген-презентацию вируса, инфицировавшего клетки КМ, и активировать CD8+ цитотоксические клетки, которые осуществляют агрессию против инфицированных гемопоэтических клеток. По данным Brown K.E. и cоавт., у пациентов с ГА-АА отмечается активация цитотоксических лимфоцитов [43]. Причем спектротипирование субсемейств Vb регионов ТКР лимфоцитов периферической крови больных ГА-АА показало антиген-специфическую экспансию CD8+ Т-клеток [42]. При ГА-АА иммуносупрессивная терапия приводит к восстановлению нормального Т-клеточного репертуара и к медленной регрессии аплазии кроветворения [42, 43]. Это является еще одним свидетельством в пользу иммунных нарушений, лежащих в основе развития ГА-АА.
Нельзя не учитывать, что обычно ДК осуществляют презентацию не только экзогенных, но и аутологичных антигенов, и в условиях отсутствия одновременного костимуляторного сигнала развивается антиген-специфическая анергия Т-лимфоцитов. Однако при сильно выраженной воспалительной реакции капсид вируса или бактериальные антигены могут послужить активационным сигналом не только для антиген-специфического взаимодействия ДК/Т-клетка, но и для процесса распознания аутоантигенов [44]. Таким образом, неинфицированные гемопоэтические клетки могут стать мишенями иммунного процесса, приводящего к аплазии апластической анемии. Подтверждением данного положения являются результаты исследований Zeng W. и соавт., которые методами обратной транскриптазной полимеразной цепной реакции и генных чипов выявили активацию как врожденного, так и адаптивного иммунных ответов при апластической анемии [4].
На основании изучения поздних осложнений апластической анемии — клональных заболеваний, развивающихся на фоне длительной ремиссии после успешной иммуносупрессивной терапии, появилось предположение о клональной природе апластической анемии. Миелодиспластический синдром с дальнейшей трансформацией в острый миелобластный лейкоз (ОМЛ) развивается в 9,6%, а ОМЛ — в 6,6% случаев апластической анемии через 5−10 лет после иммуносупрессивной терапии [8, 45]. Генетически измененные гемопоэтические клетки могут экспрессировать новые белки и, следовательно, провоцировать иммунный ответ. Однако на ранних стадиях заболевания в КМ больных апластической анемии наличие мутаций не выявлено цитогенетическими и молекулярными методами. С другой стороны, при АА снижение экспрессии c-myc, c-myb генов, контролирующих клеточный цикл, может способствовать выживанию предопухолевых и анеуплоидных клеток в КМ, особенно после иммуносупрессии, и развитию миелодиспластического синдрома и ОМЛ [4].
Пароксизмальная ночная гемоглобинурия (ПНГ), характеризующаяся мутациями гена PIG-A и дефицитом или отсутствием глюкозилфосфатидилинозитола (GPI), является еще одной клональной патологией, которая предшествует или сопутствует АА в 9−25% случаев [8, 46]. По данным некоторых авторов, клон ПНГ-дефектных клеток выявляется в 50% случаях манифестации апластической анемии [9, 47]. В течение 5−10 лет после лечения антитимоцитарным глобулином ПНГ наблюдается у 9−13% больных [48], а через 15 лет после лечения данный показатель достигает 10−25% [49]. Однако патогенетическая связь между данными заболеваниями остается неясной. Отмечено, что клон ПНГ-дефектных клеток может существовать в КМ здоровых лиц и прогрессировать в определенных условиях [8, 49]. Кроме того, пролиферативная способность GPI-дефицитных и неизмененных клеток предшественников гемопоэза одинакова при апластической анемии, сопровождающейся синдромом ПНГ [9]. Таким образом, влияние GPI-дефицитного клона на развитие аплазии кроветворения является предметом дальнейшего изучения. Доказано наличие аутоагрессивных Т-клеток с характерным изменением Vb вариабельных регионов ТКР при ПНГ. Однако некоторые поверхностные молекулы, связанные с GPI, представляют собой лиганды для рецепторов, экспрессирующихся Т-лимфоцитами, и при дефиците GPI белков клетки ПНГ оказываются более устойчивыми к апоптозу [8, 9]. Наличие множественных мутаций PIG-A гена при АА/ПНГ, в отличие от ПНГ, протекающей без аплазии кроветворения, может свидетельствовать о повышенной мутабельности гемопоэтических клеток при АА, сопровождающейся синдромом ПНГ [49].
Таким образом, гемопоэтические клетки, поврежденные химическими веществами и/или медикаментами (А), инфицированные вирусом (Б) или измененные в результате клональной трансформации (В), помимо нормальных клеточных белков экспрессируют патологические белки или вирусные антигены, которые распознаются АПК (рис. 1). ДК путем продукции цитокинов и взаимодействия молекул I или II класса ГКГС с ТКР, экспрессируемыми на поверхности Т-клеток, индуцируют Тh1-иммунный ответ. Активированные T-хелперы и цитотоксические T-клетки вызывают подавление пролиферации гемопоэтических клеток и их апоптоз.
анемия геном цитотоксический стволовой клетка.
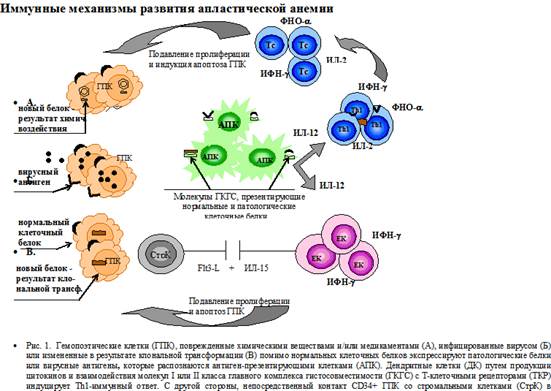
Изучение функциональных особенностей ДК при апластической анемии не проводилось, но их роль в возникновении Тh1-иммунного ответа при апластической анемии не вызывает сомнений. S. Nakao и соавт. у больных циклоспорин-А-зависимой апластической анемии, у которых была отмечена экспрессия HLA-DRB1*0405 аллели, выделили CD4+Vb21+ T-клон, способный при отсутствии экзогенного антигена подавлять рост предшественников гемопоэза, полученных после культивации CD34+ стволовых клеток в присутствие ГМ-КСФ и ИЛ-4 в течение 7 дней, но не чистой популяции CD34+ клеток [14]. Поскольку эти цитокины создают благоприятные условия для генерации популяции ДК, авторы высказали предположение, что в период культивации часть стволовых клеток дифференцировалась в ДК, которые путем презентации антигенов CD34+ стволовых клеток способствовали возникновению клона цитотоксических CD4+Vb21+ T-лимфоцитов. Данное исследование подчеркивает роль ДК в развитии АА.
HLA-DR2-специфический характер иммунного ответа против CD34+ стволовых клеток при АА доказан S. Nakao и соавт., которые анти-HLA-DR2 моноклональными антителами блокировали цитотоксичность CD4+ T-клеток, выделенных из периферических мононуклеаров больных АА [14]. Высокая частота встречаемости HLA-DR2 гаплотипов у больных АА (в 1,9 — 2 раза превышает показатель в здоровой популяции) [1, 14, 50, 51, 52, 53], также, свидетельствует об участии HLA-DR2 молекул ГКГС в презентации антигена и возникновении цитотоксического иммунного ответа при АА.
Выявлены разные гаплотипы — HLA-DRВ1*0405 и HLA-DRВ1*1501 [1, 14, 15, 47, 50], которые ассоциируются с циклоспорин-А-зависимой апластической анемии (случаи апластической анемии, при которых ремиссия достигается только после терапии циклоспорином-А и снижение дозы препарата вызывает развитие рецидива заболевания) и имеют прогностическую значимость: 71% больных апластической анемии с HLA-DRВ1*1501 гаплотипом отвечают на терапию циклоспорином-А по сравнению с 36% больных апластической анемии с HLA-DQВ1*0601 гаплотипом [50]. О подобной корреляции между гаплотипами молекул ГКГС и эффективностью терапии антитимоцитарным глобулином не сообщается.
Роль молекул ГКГС в иммуннорегуляции более сложна — кроме антиген-презентации они участвуют в возникновении центральной толерантности. Хорошо известна связь между полиморфизмом аминокислотных последовательностей HLA-DR3/HLA-DR4 гаплотипов молекул ГКГС и иммунопатологическими заболеваниями [44].
По данным S. Nakao и соавт., полиморфизма нуклеотидных последовательностей генных фрагментов характерных молекул ГКГС при АА не наблюдается [50]. Однако относительно новые исследования выявили, что 100% больных тяжелой АА экспрессировали HLA-DR4-Ала74b подтип молекул ГКГС (HLA-DRВ1*04 вариант, в 74-й позиции b-цепи которого находится аминокислота аланин), по сравнению с 62% контрольной группы здоровых доноров [52]. Авторы предполагают, что HLA-DR4-Ала74b подтип отвечает за вероятность развития тяжелой апластической анемии, и он служит крайне неблагоприятным прогностическим фактором [52]. Полиморфизм аминокислотной последовательности влияет на прочность и стабильность структуры HLA-DR молекулы.
Следовательно, молекулы HLA-DR4-Ала74b не способны образовывать высокоафинные связи с ТКР, что препятствует негативной селекции аутоагрессивной популяции Т-клеток в тимусе.
Кроме того, высокоафинные связи молекул II класса ГКГС, присутствующих на эпителиоцитах тимуса, с Т-клеточными рецепторами играют важную роль в развитии CD4+CD25+ популяции регуляторных Т-клеток в тимусе [54]. Нарушение центральной толерантности может создать предпосылки для возникновения иммунопатологического процесса, который при соответствующей антигенной стимуляции способен привести к специфической деструкции клеток КМ.
Иммуннорегуляторное звено, предотвращающее развитие нежелательного иммунного ответа, остается не изученным при данном заболевании. Дефицит иммуносупрессивных цитокинов (ТФР-в1, ИЛ-10) и разных популяций регуляторных Т-клеток может служить дополнительным фактором для усугубления иммунного процесса при апластической анемии.
ТФР-в1 является важным иммунорегуляторным цитокином, который подавляет созревание ДК, активацию T-bet и GATA3 факторов транскрипции и, следовательно, индукцию Тh1 и Тh2 иммунных ответов (30), а также раннюю пролиферацию ЕК [55]. Кроме того, ТФР-в1 in vitro блокирует Fas-опосредованный апоптоз гемопоэтических предшественников [56, 57]. Так как для апластической анемии характерны преобладание Тh1 иммунного ответа и повышенная экспрессия Fas-рецепторов в результате влияния ИФН-g и ФНО-б, дефицит данного иммуносупрессивного фактора может способствовать развитию АА.
Единичные исследования показали значительное снижение продукции трансформирующего фактора роста-в1 (ТФР-в1) у данной категории больных. Taketazu и соавт. показали снижение спонтанной продукции ТФР-в1 лимфоцитами больных апластической анемии in vitro и высказали предположение, что при этом также снижается продукция тРНК данного цитокина [58].
По данным S. Rizzo и соавт. у больных апластической анемии выявляется низкий уровень ТФР-в1 в сыворотке, что коррелирует с глубиной тромбоцитопении, а также значительно сниженная секреция ТФР-в1 стромальными клетками КМ in vitro, по сравнению со здоровыми лицами [56].
Исследование особенностей продукции ИЛ-10 у больных апластической анемии не проводилось. Единичные экспериментальные исследования показали, что ИЛ-10 эффективно подавляет супрессию гемопоэтических предшественников мононуклеарными клетками периферической крови больных апластической анемии, и высказали предположение о полезности применения ИЛ-10 в терапевтических целях у некоторых больных апластической анемии [59, 60].
В заключении следует отметить, что остается неизвестным, является ли иммунопатологический процесс при апластической анемии результатом иммунного ответа против постоянно присутствующего иммуногенного агента в качестве дефектных стволовых клеток или следствием нарушения равновесия взаимодействий различных иммуно-регуляторных механизмов. В этой связи дальнейшее изучение функциональных особенностей антиген-презентирующих клеток и нарушений регуляторного звена иммунной системы при апластической анемии можно считать актуальным.